
Comparative Transcriptome Analysis Revealed Key Genes Regulating Gossypol Synthesis in Tetraploid Cultivated Cotton
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction, Library Construction, and RNA Sequencing
2.3. RNA-seq Data Analysis
2.4. Construction of Gene Co-Expression Networks
2.5. Function Annotation of DEGs
2.6. Validation of DEGs by qRT-PCR
3. Results
3.1. Transcriptome Analysis
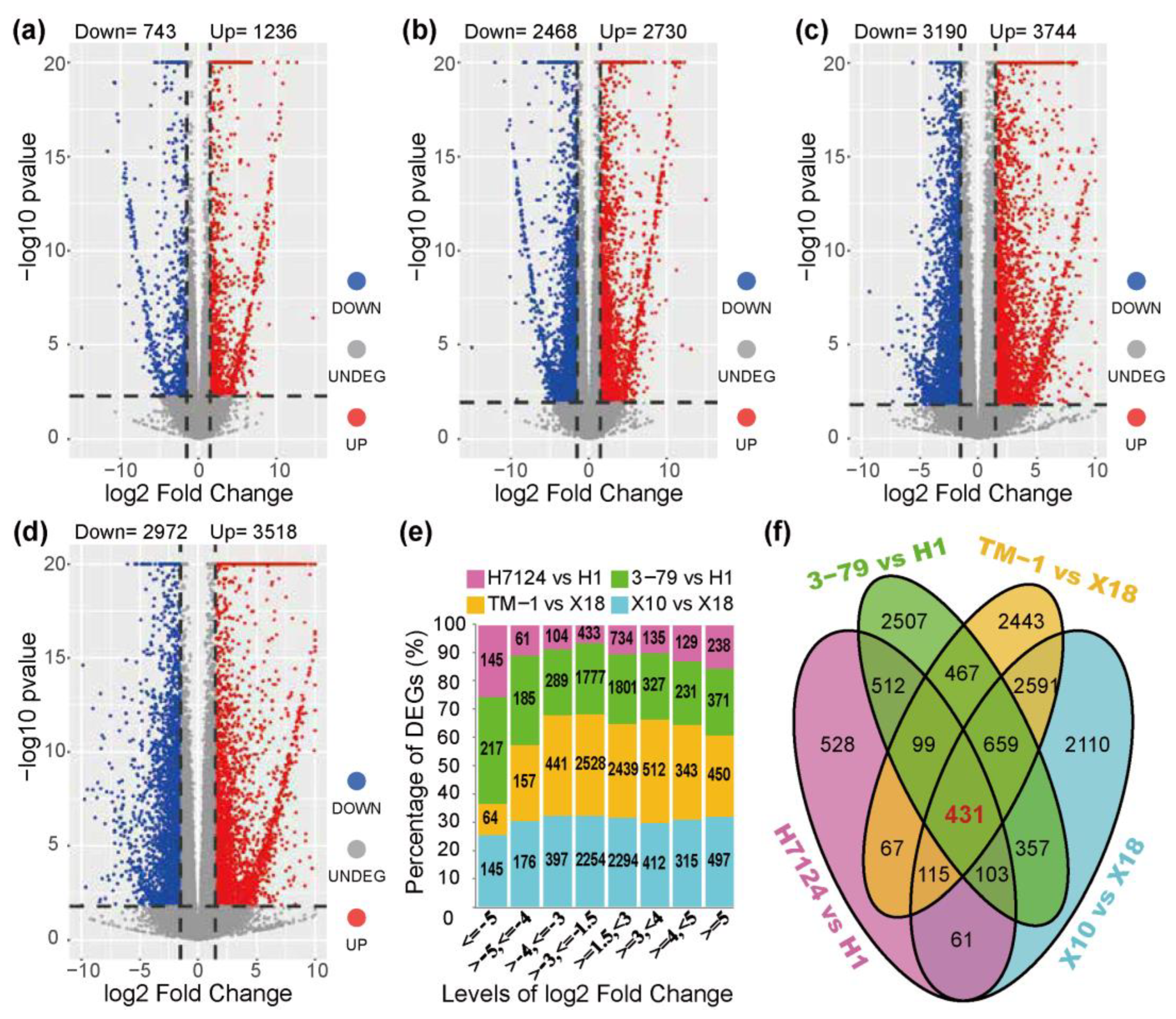
3.2. Identification of DEGs
3.3. WGCNA Analysis of DEGs
3.4. Construction of Co-Expression Gene Networks and Identification of Hub Genes
3.5. Functional Annotations of DEGs in Candidate Module
3.6. Confirmation of DEGs by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sunilkumar, G.; Campbell, L.M.; Puckhaber, L.; Stipanovic, R.D.; Rathore, K.S. Engineering cottonseed for use in human nutrition by tissue-specific reduction of toxic gossypol. Proc. Natl. Acad. Sci. USA 2006, 103, 18054–18059. [Google Scholar] [CrossRef]
- Janga, M.R.; Pandeya, D.; Campbell, L.M.; Konganti, K.; Villafuerte, S.T.; Puckhaber, L.; Pepper, A.; Stipanovic, R.D.; Scheffler, J.A.; Rathore, K.S. Genes regulating gland development in the cotton plant. Plant Biotechnol. J. 2019, 17, 1142–1153. [Google Scholar] [CrossRef]
- Gao, W.; Xu, F.C.; Long, L.; Li, Y.; Zhang, J.L.; Chong, L.; Botella, J.R.; Song, C.P. The gland localized CGP1 controls gland pigmentation and gossypol accumulation in cotton. Plant Biotechnol. J. 2020, 18, 1573–1584. [Google Scholar] [CrossRef]
- Gao, W.; Long, L.; Zhu, L.-F.; Xu, L.; Gao, W.-H.; Sun, L.-Q.; Liu, L.-L.; Zhang, X.-L. Proteomic and virus-induced gene silencing (VIGS) analyses reveal that gossypol, brassinosteroids, and jasmonic acid contribute to the resistance of cotton to Verticillium dahliae. Mol. Cell. Proteom. 2013, 12, 3690–3703. [Google Scholar] [CrossRef]
- Tian, X.; Ruan, J.-X.; Huang, J.-Q.; Yang, C.-Q.; Fang, X.; Chen, Z.-W.; Hong, H.; Wang, L.-J.; Mao, Y.-B.; Lu, S.; et al. Characterization of gossypol biosynthetic pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E5410–E5418. [Google Scholar] [CrossRef]
- Benedict, C.R.; Martin, G.S.; Liu, J.; Puckhaber, L.; Magill, C.W. Terpenoid aldehyde formation and lysigenous gland storage sites in cotton: Variant with mature glands but suppressed levels of terpenoid aldehydes. Phytochemistry 2004, 65, 1351–1359. [Google Scholar] [CrossRef]
- Townsend, B.J.; Poole, A.; Blake, C.J.; Llewellyn, D.J. Antisense suppression of a (+)-delta-cadinene synthase gene in cotton prevents the induction of this defense response gene during bacterial blight infection but not its constitutive expression. Plant Physiol. 2005, 138, 516–528. [Google Scholar] [CrossRef]
- Singh, I.D.; Weaver, J.B., Jr. Growth and infestation of boll weevils on normal-glanded, glandless, and high-gossypol strains of cotton. J. Econ. Entomol. 1972, 65, 821–824. [Google Scholar] [CrossRef]
- Wilson, F.; Smith, J. Some genetic relationships between gland density and gossypol content in Gossypium hirsutum L. Crop Sci. 1976, 16, 830–832. [Google Scholar] [CrossRef]
- Mohan, P.; Singh, P.; Dongre, A.B.; Narayanan, S.S. Gossypol-gland density and free gossypol content in seed and cotyledonary leaf of upland cotton (Gossypium hirsutum). Indian J. Agr. Sci. 1995, 65, 66–68. [Google Scholar]
- Zhao, T.; Xie, Q.; Li, C.; Li, C.; Mei, L.; Yu, J.Z.; Chen, J.; Zhu, S. Cotton roots are the major source of gossypol biosynthesis and accumulation. BMC Plant Biol. 2020, 20, 88. [Google Scholar] [CrossRef]
- Fryxell, P.A. A redefinition of the tribe Gossypieae. Bot. Gaz. 1968, 129, 296–308. [Google Scholar] [CrossRef]
- Tong, X.; Zhu, S.; Ji, D. The expression and anatomical observation of the delayed pigment gland morphogenesis in an upland cotton germplasm. Cotton Sci 2005, 17, 137–140. [Google Scholar]
- Fulton, H.J. Hopi cotton: A variable species. J. Agr. Res. 1938, 56, 333–336. [Google Scholar]
- McMichael, S. Glandless boll in upland cotton and its use on the study of natural crossing. Agron. J. 1954, 46, 527–528. [Google Scholar] [CrossRef]
- McMichael, S.C. Hopi cotton, a source of cottonseed free of gossypol pigments. Agron. J. 1959, 51, 630. [Google Scholar] [CrossRef]
- McMichael, S.C. Combined effects of the glandless gene gl2 and gl3 on pigment glands in the cotton plant. Agron. J. 1960, 52, 385–386. [Google Scholar] [CrossRef]
- Endrizzi, J.E.; Turcotte, E.L.; Kohel, R.J. Genetics, cytology, and evolution of Gossypium. Adv. Genet. 1985, 23, 271–375. [Google Scholar] [CrossRef]
- Lee, J.A. The genomic allocation of the principal foliar-gland loci in Gossypium hirsutum and Gossypium barbadense. Evolution 1965, 19, 182–188. [Google Scholar] [CrossRef]
- Kohel, R.J.; Lee, J.A. Genetic analysis of Egyptian glandless seeds cotton. Crop Sci. 1984, 24, 1119–1121. [Google Scholar] [CrossRef]
- Tang, C.; Min, L.; Zhang, T.; Pan, J.; Jing, S.; Yuan, Y.; Liu, S. Genetic analysis for Hai-1 strain of glandless cotton (G. barbadence L.) interaction between Gl2e and Gl1. Cotton Sci. Sin 1996, 8, 138–140. [Google Scholar]
- Afifi, A.; Bary, A.; Kamel, S.; Heikal, I. Bahtim 110, a new strain of Egyptian cotton free from gossypol. Emp. Cot. Grow. Rew 1966, 43, 112–120. [Google Scholar]
- Cheng, H.; Lu, C.; Yu, J.Z.; Zou, C.; Zhang, Y.; Wang, Q.; Huang, J.; Feng, X.; Jiang, P.; Yang, W.; et al. Fine mapping and candidate gene analysis of the dominant glandless gene Gl2e in cotton (Gossypium spp.). Theor. Appl. Genet. 2016, 129, 1347–1355. [Google Scholar] [CrossRef]
- Ma, D.; Hu, Y.; Yang, C.; Liu, B.; Fang, L.; Wan, Q.; Liang, W.; Mei, G.; Wang, L.; Wang, H.; et al. Genetic basis for glandular trichome formation in cotton. Nat. Commun. 2016, 7, 10456. [Google Scholar] [CrossRef]
- Zang, Y.; Xu, C.; Xuan, L.; Ding, L.; Zhu, J.; Si, Z.; Zhang, T.; Hu, Y. Identification and characteristics of a novel gland-forming gene in cotton. Plant J. 2021, 108, 781–792. [Google Scholar] [CrossRef]
- Cai, Y.; Cai, X.; Wang, Q.; Wang, P.; Zhang, Y.; Cai, C.; Xu, Y.; Wang, K.; Zhou, Z.; Wang, C.; et al. Genome sequencing of the Australian wild diploid species Gossypium australe highlights disease resistance and delayed gland morphogenesis. Plant Biotechnol. J. 2020, 18, 814–828. [Google Scholar] [CrossRef]
- El-Sharkawy, I.; Liang, D.; Xu, K. Transcriptome analysis of an apple (Malus × domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J. Exp. Bot. 2015, 66, 7359–7376. [Google Scholar] [CrossRef]
- Deng, T.; Liang, A.; Liang, S.; Ma, X.; Lu, X.; Duan, A.; Pang, C.; Hua, G.; Liu, S.; Campanile, G.; et al. Integrative analysis of transcriptome and GWAS data to identify the hub genes associated with milk yield trait in buffalo. Front. Genet. 2019, 10, 36. [Google Scholar] [CrossRef]
- Cheng, X.-Q.; Zhang, X.-Y.; Xue, F.; Zhu, S.-H.; Li, Y.-J.; Zhu, Q.-H.; Liu, F.; Sun, J. Characterization and transcriptome analysis of a dominant genic male sterile cotton mutant. BMC Plant Biol. 2020, 20, 312. [Google Scholar] [CrossRef]
- Han, Z.; Ahsan, M.; Adil, M.F.; Chen, X.; Nazir, M.M.; Shamsi, I.H.; Zeng, F.; Zhang, G. Identification of the gene network modules highly associated with the synthesis of phenolics compounds in barley by transcriptome and metabolome analysis. Food Chem. 2020, 323, 126862. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 2008, 9, 559. [Google Scholar] [CrossRef]
- Li, S.; Geng, S.; Pang, B.; Zhao, J.; Huang, Y.; Rui, C.; Cui, J.; Jiao, Y.; Zhang, R.; Gao, W. Revealing genetic differences in fiber elongation between the offspring of sea island cotton and upland cotton backcross populations based on transcriptome and weighted gene coexpression networks. Genes 2022, 13, 954. [Google Scholar] [CrossRef]
- Sun, S.; Xiong, X.-p.; Zhu, Q.; Li, Y.-j.; Sun, J. Transcriptome sequencing and metabolome analysis reveal genes involved in pigmentation of green-colored cotton fibers. Int. J. Mol. Sci. 2019, 20, 4838. [Google Scholar] [CrossRef]
- Zou, X.; Liu, A.; Zhang, Z.; Ge, Q.; Fan, S.; Gong, W.; Li, J.; Gong, J.; Shi, Y.; Tian, B.; et al. Co-expression network analysis and hub gene selection for high-quality fiber in upland cotton (Gossypium hirsutum) using RNA sequencing analysis. Genes 2019, 10, 119. [Google Scholar] [CrossRef]
- Mehari, T.G.; Hou, Y.; Xu, Y.; Umer, M.J.; Shiraku, M.L.; Wang, Y.; Wang, H.; Peng, R.; Wei, Y.; Cai, X.; et al. Overexpression of cotton GhNAC072 gene enhances drought and salt stress tolerance in transgenic Arabidopsis. BMC Genom. 2022, 23, 648. [Google Scholar] [CrossRef]
- Jing, S.; Zhan, X. Selection of new types of dominant glandless cotton (Gossypium hirsutum) germplasm. Sci. Agric. Sin. 1990, 23, 22–27. [Google Scholar]
- Zhang, X.; Jin, L.; Zhang, T. A new upland cotton cultivar with glanded plant and low gossypol content seed. Sci. Agric. Sin. 2001, 34, 564–567. [Google Scholar]
- Zhang, T.; Zhang, X.; Jin, L.; Chen, Z.; Guo, W. Genetic identification of a new gland forming gene in upland cotton. Acta Agron. Sin. 2001, 27, 75–79. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhong, X.; Lian, D.; Zheng, Y.; Wang, H.; Liu, X. Triterpenoid biosynthesis and the transcriptional response elicited by nitric oxide in submerged fermenting Ganoderma lucidum. Process Biochem. 2017, 60, 19–26. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Rubio, V. ggplot2—Elegant graphics for data analysis (2nd Edition). J. Stat. Soft. 2017, 77, 1–3. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Liang, C.; Meng, Z.; Sun, G.; Meng, Z.; Guo, S.; Zhang, R. CottonFGD: An integrated functional genomics database for cotton. BMC Plant Biol. 2017, 17, 101. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, W. OLIGO7 primer analysis software. Methods Mol. Biol. 2007, 402, 35–59. [Google Scholar] [CrossRef]
- Smith, F.H. Biosynthesis of Gossypol by excised cotton roots. Nature 1961, 192, 888–889. [Google Scholar] [CrossRef]
- Kong, G.; Daud, M.K.; Zhu, S. Effects of pigment glands and gossypol on growth, development and insecticide-resistance of cotton bollworm (Heliothis armigera (Hübner)). Crop Protect. 2010, 29, 813–819. [Google Scholar] [CrossRef]
- Kapoor, S. Attenuating effect of gossypol on tumor growth in systemic malignancies. Cell Biochem. Biophys. 2013, 67, 1551–1552. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Huang, J.-Q.; Chen, X.-Y.; Zhu, Y.-X. Recent advances and future perspectives in cotton research. Annu. Rev. Plant Biol. 2021, 72, 437–462. [Google Scholar] [CrossRef] [PubMed]
- Sheng, K.; Sun, Y.; Liu, M.; Cao, Y.; Han, Y.; Li, C.; Muhammad, U.; Daud, M.K.; Wang, W.; Li, H.; et al. A reference-grade genome assembly for Gossypium bickii and insights into its genome evolution and formation of pigment glands and gossypol. Plant Commun. 2023, 4, 100421. [Google Scholar] [CrossRef] [PubMed]
- Zha, H.-G.; Liu, T.; Zhou, J.-J.; Sun, H. MS-desi, a desiccation-related protein in the floral nectar of the evergreen velvet bean (Mucuna sempervirens Hemsl): Molecular identification and characterization. Planta 2013, 238, 77–89. [Google Scholar] [CrossRef]
- Schmidt, M.A.; Barbazuk, W.B.; Sandford, M.; May, G.; Song, Z.; Zhou, W.; Nikolau, B.J.; Herman, E.M. Silencing of soybean seed storage proteins results in a rebalanced protein composition preserving seed protein content without major collateral changes in the metabolome and transcriptome. Plant Physiol. 2011, 156, 330–345. [Google Scholar] [CrossRef]
- Rathore, K.S.; Sundaram, S.; Sunilkumar, G.; Campbell, L.M.; Puckhaber, L.; Marcel, S.; Palle, S.R.; Stipanovic, R.D.; Wedegaertner, T.C. Ultra-low gossypol cottonseed: Generational stability of the seed-specific, RNAi-mediated phenotype and resumption of terpenoid profile following seed germination. Plant Biotechnol. J. 2012, 10, 174–183. [Google Scholar] [CrossRef]
- Palle, S.R.; Campbell, L.M.; Pandeya, D.; Puckhaber, L.; Tollack, L.K.; Marcel, S.; Sundaram, S.; Stipanovic, R.D.; Wedegaertner, T.C.; Hinze, L.; et al. RNAi-mediated Ultra-low gossypol cottonseed trait: Performance of transgenic lines under field conditions. Plant Biotechnol. J. 2013, 11, 296–304. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, J.; Sun, L.; Ma, Y.; Xu, J.; Liang, S.; Deng, J.; Tan, J.; Zhang, Q.; Tu, L.; et al. High efficient multisites genome editing in allotetraploid cotton (Gossypium hirsutum) using CRISPR/Cas9 system. Plant Biotechnol. J. 2018, 16, 137–150. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, L.; Li, S.; Qian, Y.; Cheng, H.; Zhang, Y.; Zuo, D.; Lv, L.; Wang, Q.; Li, J.; Song, G. Comparative Transcriptome Analysis Revealed Key Genes Regulating Gossypol Synthesis in Tetraploid Cultivated Cotton. Genes 2023, 14, 1144. https://doi.org/10.3390/genes14061144
Kong L, Li S, Qian Y, Cheng H, Zhang Y, Zuo D, Lv L, Wang Q, Li J, Song G. Comparative Transcriptome Analysis Revealed Key Genes Regulating Gossypol Synthesis in Tetraploid Cultivated Cotton. Genes. 2023; 14(6):1144. https://doi.org/10.3390/genes14061144
Chicago/Turabian StyleKong, Linglei, Shaoqi Li, Yuyuan Qian, Hailiang Cheng, Youping Zhang, Dongyun Zuo, Limin Lv, Qiaolian Wang, Junlan Li, and Guoli Song. 2023. "Comparative Transcriptome Analysis Revealed Key Genes Regulating Gossypol Synthesis in Tetraploid Cultivated Cotton" Genes 14, no. 6: 1144. https://doi.org/10.3390/genes14061144
APA StyleKong, L., Li, S., Qian, Y., Cheng, H., Zhang, Y., Zuo, D., Lv, L., Wang, Q., Li, J., & Song, G. (2023). Comparative Transcriptome Analysis Revealed Key Genes Regulating Gossypol Synthesis in Tetraploid Cultivated Cotton. Genes, 14(6), 1144. https://doi.org/10.3390/genes14061144