
Interaction between KLOTHO-VS Heterozygosity and APOE ε4 Allele Predicts Rate of Cognitive Decline in Late-Onset Alzheimer’s Disease

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Participant Selection
2.2. Genetic Data Quality Control and Processing
2.3. Cognitive Measures
2.4. Statistical Analyses
3. Results
3.1. Descriptive Statistics
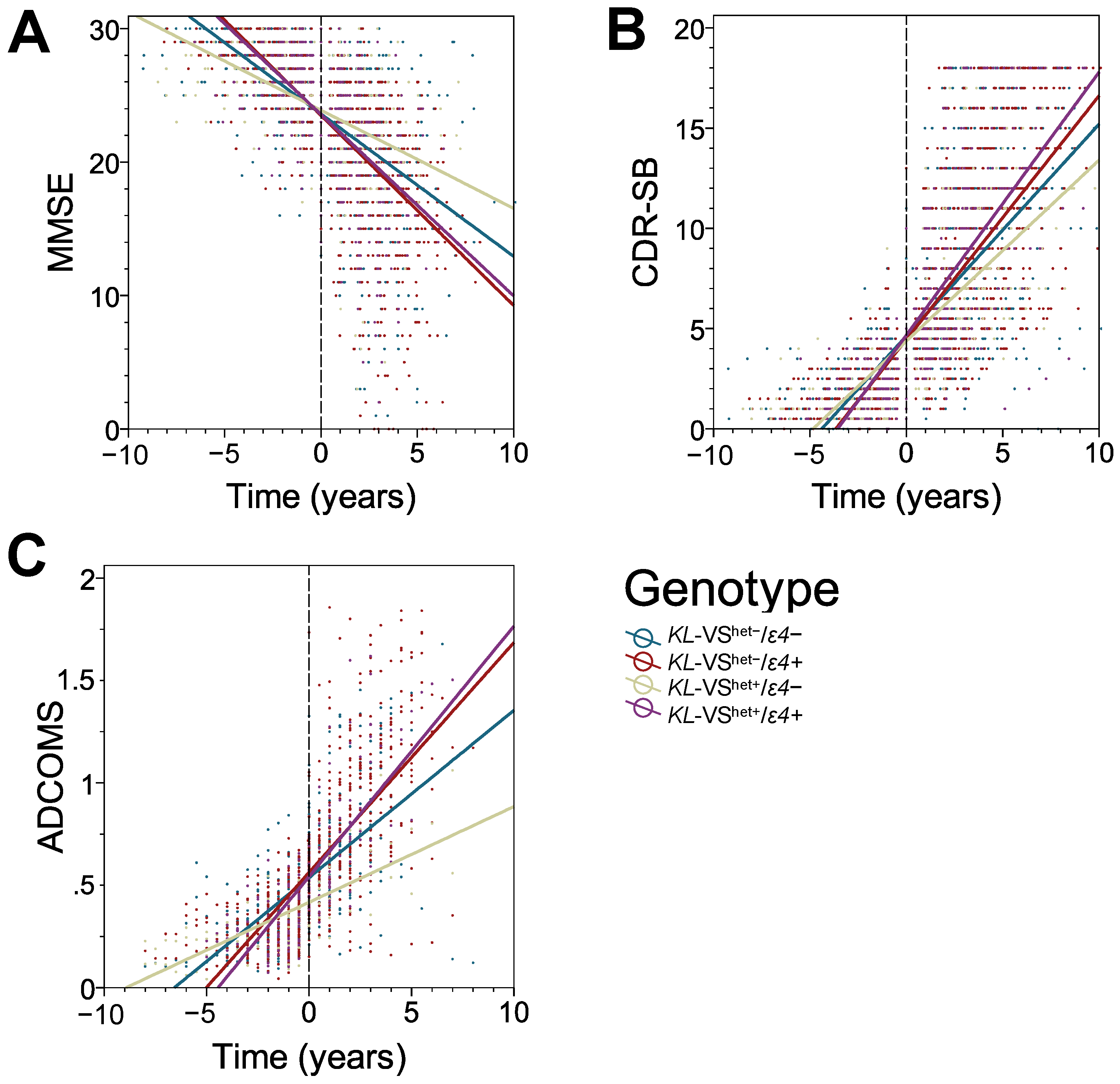
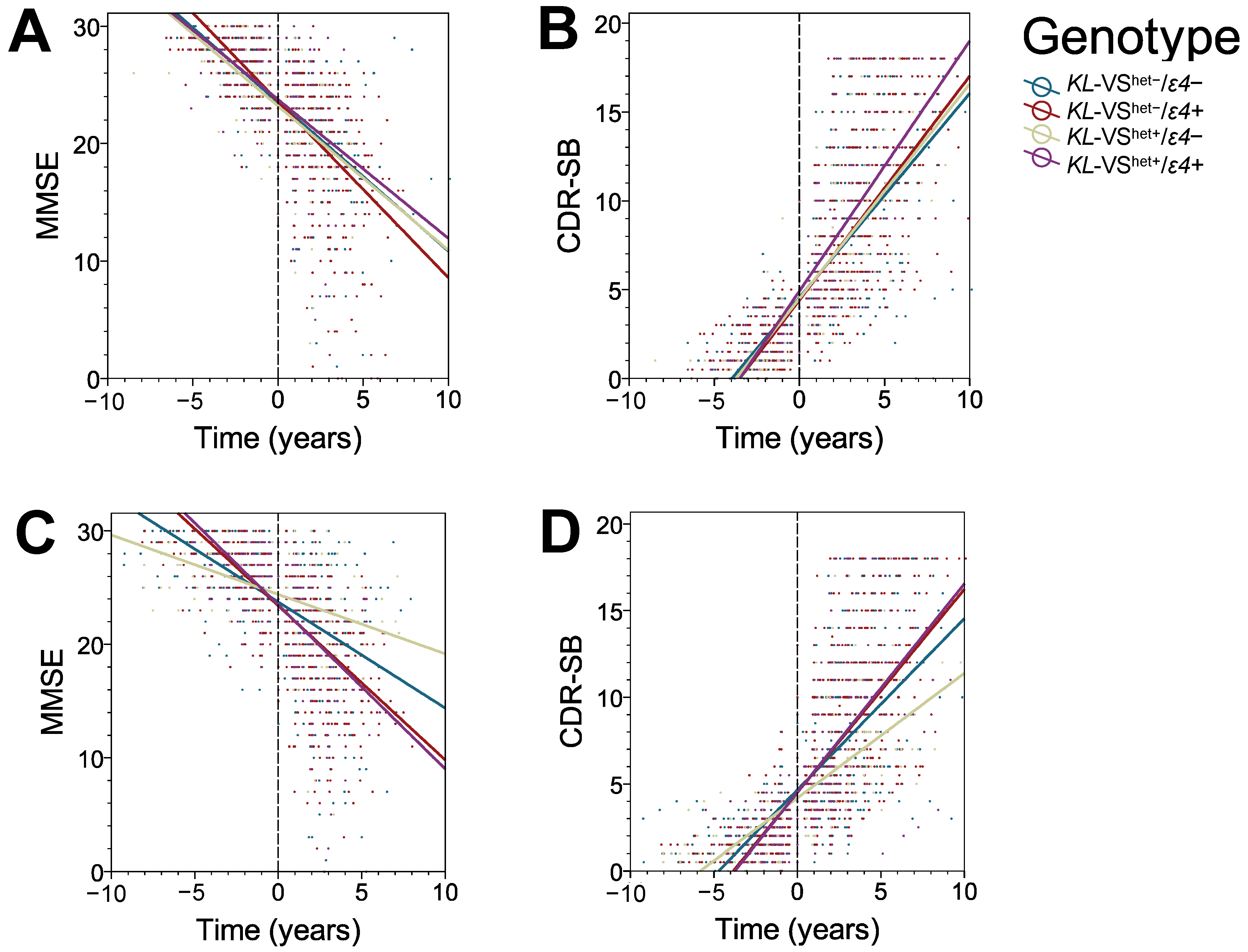
3.2. KL-VS Heterozygosity Has a Protective Effect on the Rate of Cognitive Decline in ε4 Non-Carriers
3.3. The Protective Effect of KL-VS Heterozygosity Is Observed in Males, in Individuals with an Older Age of Cognitive Decline Onset, and in Those with 16 or More Years of Education
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tohyama, O.; Imura, A.; Iwano, A.; Freund, J.N.; Henrissat, B.; Fujimori, T.; Nabeshima, Y. Klotho is a novel beta-glucuronidase capable of hydrolyzing steroid beta-glucuronides. J. Biol. Chem. 2004, 279, 9777–9784. [Google Scholar] [CrossRef] [PubMed]
- Arking, D.E.; Krebsova, A.; Macek, M., Sr.; Macek, M., Jr.; Arking, A.; Mian, I.S.; Fried, L.; Hamosh, A.; Dey, S.; McIntosh, I.; et al. Association of human aging with a functional variant of klotho. Proc. Natl. Acad. Sci. USA 2002, 99, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Di Bona, D.; Accardi, G.; Virruso, C.; Candore, G.; Caruso, C. Association of Klotho polymorphisms with healthy aging: A systematic review and meta-analysis. Rejuvenation Res. 2014, 17, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Dubal, D.B.; Yokoyama, J.S.; Zhu, L.; Broestl, L.; Worden, K.; Wang, D.; Sturm, V.E.; Kim, D.; Klein, E.; Yu, G.; et al. Life extension factor klotho enhances cognition. Cell Rep. 2014, 7, 1065–1076. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef]
- Kuro, M. Klotho as a regulator of oxidative stress and senescence. Biol. Chem. 2008, 389, 233–241. [Google Scholar] [CrossRef]
- Zeldich, E.; Chen, C.D.; Colvin, T.A.; Bove-Fenderson, E.A.; Liang, J.; Tucker-Zhou, T.B.; Harris, D.A.; Abraham, C.R. The neuroprotective effect of Klotho is mediated via regulation of members of the redox system. J. Biol. Chem. 2014, 289, 24700–24715. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; Abdoli, A.; Abualhasan, A.; Abu-Gharbieh, E.; Akram, T.T.; et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Rajan, K.B.; Weuve, J.; Barnes, L.L.; McAninch, E.A.; Wilson, R.S.; Evans, D.A. Population estimate of people with clinical Alzheimer’s disease and mild cognitive impairment in the United States (2020–2060). Alzheimer Dement. 2021, 17, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Sclan, S.G.; Reisberg, B. Functional Assessment Staging (FAST) in Alzheimer’s disease: Reliability, validity and ordinality. Int. Psychogeriatr. 1992, 4, 55–69. [Google Scholar]
- Petersen, R.C.; Stevens, J.C.; Ganguli, M.; Tangalos, E.G.; Cummings, J.L.; DeKosky, S.T. Practice parameter: Early detection of dementia: Mild cognitive impairment (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2001, 56, 1133–1142. [Google Scholar] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [PubMed]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar]
- Song, S.; Stern, Y.; Gu, Y. Modifiable lifestyle factors and cognitive reserve: A systematic review of current evidence. Ageing Res. Rev. 2022, 74, 101551. [Google Scholar] [PubMed]
- Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012, 11, 1006–1012. [Google Scholar]
- Neitzel, J.; Franzmeier, N.; Rubinski, A.; Dichgans, M.; Brendel, M.; Alzheimer’s Disease Neuroimaging Initiative (ADNI); Malik, R.; Ewers, M. KL-VS heterozygosity is associated with lower amyloid-dependent tau accumulation and memory impairment in Alzheimer’s disease. Nat. Commun. 2021, 12, 3825. [Google Scholar] [PubMed]
- Driscoll, I.; Ma, Y.; Gallagher, C.L.; Johnson, S.C.; Asthana, S.; Hermann, B.P.; Sager, M.A.; Blennow, K.; Zetterberg, H.; Carlsson, C.M.; et al. Age-Related Tau Burden and Cognitive Deficits Are Attenuated in KLOTHO KL-VS Heterozygotes. J. Alzheimer Dis. 2021, 79, 1297–1305. [Google Scholar]
- Morris, J.C.; Weintraub, S.; Chui, H.C.; Cummings, J.; Decarli, C.; Ferris, S.; Foster, N.L.; Galasko, D.; Graff-Radford, N.; Peskind, E.R.; et al. The Uniform Data Set (UDS): Clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis. Assoc. Disord. 2006, 20, 210–216. [Google Scholar] [PubMed]
- Weiner, M.W.; Aisen, P.S.; Jack, C.R., Jr.; Jagust, W.J.; Trojanowski, J.Q.; Shaw, L.; Saykin, A.J.; Morris, J.C.; Cairns, N.; Beckett, L.A.; et al. The Alzheimer’s disease neuroimaging initiative: Progress report and future plans. Alzheimer Dement. 2010, 6, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.R.; Shao, Y.; Sadowski, M.J.; Alzheimer’s Disease Neuroimaging Initiative. Segmented Linear Mixed Model Analysis Reveals Association of the APOEɛ4 Allele with Faster Rate of Alzheimer’s Disease Dementia Progression. J. Alzheimer Dis. 2021, 82, 921–937. [Google Scholar] [CrossRef]
- Cosentino, S.; Scarmeas, N.; Helzner, E.; Glymour, M.M.; Brandt, J.; Albert, M.; Blacker, D.; Stern, Y. APOE epsilon 4 allele predicts faster cognitive decline in mild Alzheimer disease. Neurology 2008, 70, 1842–1849. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoΕ4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Belloy, M.E.; Napolioni, V.; Han, S.S.; Le Guen, Y.; Greicius, M.D. Association of Klotho-VS Heterozygosity with Risk of Alzheimer Disease in Individuals Who Carry APOE4. JAMA Neurol. 2020, 77, 849–862. [Google Scholar]
- Tank, R.; Ward, J.; Celis-Morales, C.; Smith, D.J.; Flegal, K.E.; Lyall, D.M. Testing for Interactions Between APOE and Klotho Genotypes on Cognitive, Dementia, and Brain Imaging Metrics in UK Biobank. J. Alzheimer Dis. 2021, 83, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [PubMed]
- Wang, J.; Logovinsky, V.; Hendrix, S.B.; Stanworth, S.H.; Perdomo, C.; Xu, L.; Dhadda, S.; Do, I.; Rabe, M.; Luthman, J.; et al. ADCOMS: A composite clinical outcome for prodromal Alzheimer’s disease trials. J. Neurol. Neurosurg. Psychiatry 2016, 87, 993–999. [Google Scholar]
- Weisgraber, K.H.; Rall, S.C., Jr.; Mahley, R.W. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J. Biol. Chem. 1981, 256, 9077–9083. [Google Scholar] [CrossRef]
- Frieden, C.; Wang, H.; Ho, C.M.W. A mechanism for lipid binding to apoE and the role of intrinsically disordered regions coupled to domain-domain interactions. Proc. Natl. Acad. Sci. USA 2017, 114, 6292–6297. [Google Scholar] [CrossRef]
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2021, 109, 205–221. [Google Scholar] [CrossRef]
- Di Battista, A.M.; Heinsinger, N.M.; Rebeck, G.W. Alzheimer’s disease genetic risk factor APOE-ε4 also affects normal brain function. Curr. Alzheimer Res. 2016, 13, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar]
- Shiozaki, M.; Yoshimura, K.; Shibata, M.; Koike, M.; Matsuura, N.; Uchiyama, Y.; Gotow, T. Morphological and biochemical signs of age-related neurodegenerative changes in klotho mutant mice. Neuroscience 2008, 152, 924–941. [Google Scholar] [CrossRef] [PubMed]
- Uchida, A.; Komiya, T.; Tashiro, T.; Yorifuji, H.; Kishimoto, T.; Nabeshima, Y.; Hisanaga, S. Neurofilaments of klotho, the mutant mouse prematurely displaying symptoms resembling human aging. J. Neurosci. Res. 2001, 64, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Yamada, K.; Kim, H.C.; Kim, Y.S.; Noda, Y.; Imura, A.; Nabeshima, Y.; Nabeshima, T. Cognition impairment in the genetic model of aging klotho gene mutant mice: A role of oxidative stress. FASEB J. 2003, 17, 50–52. [Google Scholar] [CrossRef] [PubMed]
- Nuriel, T.; Angulo, S.L.; Khan, U.; Ashok, A.; Chen, Q.; Figueroa, H.Y.; Emrani, S.; Liu, L.; Herman, M.; Barrett, G.; et al. Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat. Commun. 2017, 8, 1464. [Google Scholar] [CrossRef]
- Li, G.; Bien-Ly, N.; Andrews-Zwilling, Y.; Xu, Q.; Bernardo, A.; Ring, K.; Halabisky, B.; Deng, C.; Mahley, R.W.; Huang, Y. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell 2009, 5, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Knoferle, J.; Yoon, S.Y.; Walker, D.; Leung, L.; Gillespie, A.K.; Tong, L.M.; Bien-Ly, N.; Huang, Y. Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. J. Neurosci. 2014, 34, 14069–14078. [Google Scholar] [CrossRef]
- Chen, Y.; Durakoglugil, M.S.; Xian, X.; Herz, J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc. Natl. Acad. Sci. USA 2010, 107, 12011–12016. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef]
- Abraham, C.R.; Mullen, P.C.; Tucker-Zhou, T.; Chen, C.D.; Zeldich, E. Klotho Is a Neuroprotective and Cognition-Enhancing Protein. Vitam. Horm. 2016, 101, 215–238. [Google Scholar] [PubMed]
- Degaspari, S.; Tzanno-Martins, C.B.; Fujihara, C.K.; Zatz, R.; Branco-Martins, J.P.; Viel, T.A.; Buck, H.S.; Orellana, A.M.M.; Böhmer, A.E.; de Sá Lima, L. Altered KLOTHO and NF-κB-TNF-α Signaling Are Correlated with Nephrectomy-Induced Cognitive Impairment in Rats. PLoS ONE 2015, 10, e0125271. [Google Scholar] [CrossRef]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 412–425. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Therriault, J.; Benedet, A.L.; Pascoal, T.A.; Mathotaarachchi, S.; Chamoun, M.; Savard, M.; Thomas, E.; Kang, M.S.; Lussier, F.; Tissot, C.; et al. Association of Apolipoprotein E ε4 With Medial Temporal Tau Independent of Amyloid-β. JAMA Neurol. 2020, 77, 470–479. [Google Scholar] [CrossRef]
- Therriault, J.; Benedet, A.L.; Pascoal, T.A.; Mathotaarachchi, S.; Savard, M.; Chamoun, M.; Thomas, E.; Kang, M.S.; Lussier, F.; Tissot, C.; et al. APOEε4 potentiates the relationship between amyloid-β and tau pathologies. Mol. Psychiatry 2021, 26, 5977–5988. [Google Scholar] [CrossRef]
- Yan, S.; Zhen, C.; Paranjpe, M.; Li, Y.; Li, W.; Wang, X.; Benzinger, T.; Lu, J.; Zhou, Y. Sex modifies APOE ε4 dose effect on brain tau deposition in cognitively impaired individuals. Brain 2021, 144, 3201–3211. [Google Scholar] [CrossRef]
- Shi, Y.; Manis, M.; Long, J.; Wang, K.; Sullivan, P.M.; Serrano, J.R.; Hoyle, R.; Holtzman, D.M. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med. 2019, 216, 2546–2561. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, A.; Sung, Y.J.; Wang, F.; Fernández, M.V.; Morris, J.C.; Fagan, A.M.; Blennow, K.; Zetterberg, H.; Heslegrave, A.; Johansson, P.M.; et al. Leveraging large multi-center cohorts of Alzheimer disease endophenotypes to understand the role of Klotho heterozygosity on disease risk. PLoS ONE 2022, 17, e0267298. [Google Scholar]
- Fernández, Á.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.; et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 1–18. [Google Scholar]
- Simonovitch, S.; Schmukler, E.; Bespalko, A.; Iram, T.; Frenkel, D.; Holtzman, D.M.; Masliah, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Impaired Autophagy in APOE4 Astrocytes. J. Alzheimer Dis. 2016, 51, 915–927. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Imura, A.; Urakawa, I.; Shimada, T.; Murakami, J.; Aono, Y.; Hasegawa, H.; Yamashita, T.; Nakatani, K.; Saito, Y.; et al. Establishment of a sandwich ELISA for soluble alpha-Klotho measurements: Age-dependent change of soluble alpha-Klotho levels in healthy subjects. Biochem. Biophys. Res. Commun. 2010, 398, 513–518. [Google Scholar] [CrossRef]
- Semba, R.D.; Moghekar, A.R.; Hu, J.; Sun, K.; Turner, R.; Ferrucci, L.; O’Brien, R. Klotho in the cerebrospinal fluid of adults with and without Alzheimer’s disease. Neurosci. Lett. 2014, 558, 37–40. [Google Scholar] [CrossRef]
- Buckley, R.F.; Mormino, E.C.; Rabin, J.S.; Hohman, T.J.; Landau, S.; Hanseeuw, B.J.; Jacobs, H.I.L.; Papp, K.V.; Amariglio, R.E.; Properzi, M.J.; et al. Sex differences in the association of global amyloid and regional tau deposition measured by positron emission tomography in clinically normal older adults. JAMA Neurol. 2019, 76, 542–551. [Google Scholar] [CrossRef]
- Liu, M.; Paranjpe, M.D.; Zhou, X.; Duy, P.Q.; Goyal, M.S.; Benzinger, T.L.S.; Lu, J.; Wang, R.; Zhou, Y. Sex modulates the ApoE epsilon4 effect on brain tau deposition measured by (18)F-AV-1451 PET in individuals with mild cognitive impairment. Theranostics 2019, 9, 4959–4970. [Google Scholar] [CrossRef] [PubMed]
- Holland, D.; Desikan, R.S.; Dale, A.M.; McEvoy, L.K.; Alzheimer’s Disease Neuroimaging Initiative. Higher rates of decline for women and apolipoprotein E epsilon4 carriers. AJNR Am. J. Neuroradiol. 2013, 34, 2287–2293. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, X.; Chaput, D.; Shin, M.; Koh, Y.; Gan, L.; Pieper, A.A.; Woo, J.A.; Kang, D.E. X-linked ubiquitin-specific peptidase 11 increases tauopathy vulnerability in women. Cell 2022, 185, 3913–3930. [Google Scholar] [CrossRef]
- Mosconi, L.; Berti, V.; Quinn, C.; McHugh, P.; Petrongolo, G.; Varsavsky, I.; Osorio, R.S.; Pupi, A.; Vallabhajosula, S.; Isaacson, R.S.; et al. Sex differences in Alzheimer risk: Brain imaging of endocrine vs chronologic aging. Neurology 2017, 89, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, A.; Grundman, M.; Jack, C.R., Jr.; Petersen, R.C.; Taylor, C.; Kim, H.T.; Schiller, D.H.; Bagwell, V.; Sencakova, D.; Weiner, M.F.; et al. Sex, apolipoprotein E epsilon 4 status, and hippocampal volume in mild cognitive impairment. Arch. Neurol. 2005, 62, 953–957. [Google Scholar] [CrossRef]
- Hobel, Z.; Isenberg, A.L.; Raghupathy, D.; Mack, W.; Pa, J.; Alzheimer’s Disease Neuroimaging Intiative; Australian Imaging Biomarkers and Lifestyle Flagship Study of Ageing. APOEε4 gene dose and sex effects on Alzheimer’s disease MRI biomarkers in older adults with mild cognitive impairment. J Alzheimer Dis. 2019, 71, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.R.; He, Y.; Tekin, S.; Xu, J.; Lane, R.; Charles, H.C. Impact of APOE in mild cognitive impairment. Neurology 2004, 63, 1898–1901. [Google Scholar] [CrossRef]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef]
- Wei, X.; Yu, J.; Tan, M.; Tan, L. Cognitive reserve and Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 187–208. [Google Scholar]
- Belloy, M.E.; Eger, S.J.; Le Guen, Y.; Napolioni, V.; Deters, K.D.; Yang, H.; Scelsi, M.A.; Porter, T.; James, S.; Wong, A.; et al. KL∗VS heterozygosity reduces brain amyloid in asymptomatic at-risk APOE∗4 carriers. Neurobiol. Aging 2021, 101, 123–129. [Google Scholar] [CrossRef]
- Li, H.; Wang, B.; Wang, Z.; Guo, Q.; Tabuchi, K.; Hammer, R.E.; Südhof, T.C.; Zheng, H. Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. Proc. Natl. Acad. Sci. USA 2010, 107, 17362–17367. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Parameter | All (n = 665) | KL-VShet−/ε4− (n = 208) | KL-VShet−/ε4+ (n = 307) | KL-VShet+/ε4− (n = 66) | KL-VShet+/ε4+ (n = 84) |
|---|---|---|---|---|---|
| Number of visits | 10.7 (4.1) | 10.1 (3.8) | 11.1 (4.0) | 12.1 (5.9) | 10.0 (4.3) |
| Baseline age (years) *** | 75.5 (8.1) | 77.9 (8.8) | 74.0 (7.1) | 77.7 (8.7) | 73.2 (7.2) |
| Transition age (years) *** | 77.7 (8.2) | 80.1 (8.9) | 76.1 (7.1) | 80.3 (9.3) | 75.3 (7.4) |
| Education (years) | 16.2 (6.3) | 16.8 (8.7) | 15.8 (2.8) | 15.6 (2.9) | 16.8 (9.4) |
| % Male | 53.4% | 55.8% | 51.8% | 48.5% | 57.1% |
| APOE ε4− (p = 0.794) | APOE ε4+ (p = 0.792) | |||
|---|---|---|---|---|
| APOEGenotype | KL-VShet− | KL-VShet+ | KL-VShet− | KL-VShet+ |
| ε2/ε2 | 0 (0%) | 0 (0%) | - | - |
| ε2/ε3 | 16 (7.7%) | 6 (9.1%) | - | - |
| ε3/ε3 | 193 (92.3%) | 60 (90.9%) | - | - |
| ε2/ε4 | - | - | 12 (3.9%) | 3 (3.6%) |
| ε3/ε4 | - | - | 221 (72.2%) | 64 (76.2%) |
| ε4/ε4 | - | - | 73 (23.9%) | 17 (20.2%) |
| Cognitive Measure | Factor | β (SE) | p † |
|---|---|---|---|
| MMSE | Time VShet−/ε4− v. VShet−/ε4+ VShet+/ε4− v. VShet−/ε4− VShet+/ε4+ v. VShet−/ε4− VShet+/ε4− v. VShet−/ε4+ VShet+/ε4+ v. VShet−/ε4+ VShet+/ε4+ v. VShet+/ε4− VShet−/ε4− v. VShet−/ε4+ x Time VShet+/ε4− v. VShet−/ε4− x Time VShet+/ε4+ v. VShet−/ε4− x Time VShet+/ε4− v. VShet−/ε4+ x Time VShet+/ε4+ v. VShet−/ε4+ x Time VShet+/ε4+ v. VShet+/ε4− x Time | −1.535 (0.007) +0.169 (0.276) +0.027 (0.429) +0.027 (0.393) +0.197 (0.415) +0.196 (0.368) −0.001 (0.501) +0.290 (0.055) +0.287 (0.089) −0.325 (0.085) +0.577 (0.000) +0.036 (0.080) −0.613 (0.106) | 0.000 0.540 0.949 0.945 0.635 0.594 0.999 0.000 0.001 0.000 0.000 0.659 0.000 |
| CDR-SB | Time VShet−/ε4− v. VShet−/ε4+ VShet+/ε4− v. VShet−/ε4− VShet+/ε4+ v. VShet−/ε4− VShet+/ε4+ v. VShet−/ε4− VShet+/ε4+ v. VShet−/ε4+ VShet+/ε4+ v. VShet+/ε4− VShet−/ε4− v. VShet−/ε4+ x Time VShet+/ε4− v. VShet−/ε4− x Time VShet+/ε4+ v. VShet−/ε4− x Time VShet+/ε4− v. VShet−/ε4+ x Time VShet+/ε4+ v. VShet−/ε4+ x Time VShet+/ε4+ v. VShet+/ε4− x Time | +1.352 (0.040) −0.013 (0.187) −0.131 (0.286) +0.282 (0.267) −0.144 (0.276) +0.269 (0.251) +0.413 (0.336) −0.167 (0.031) −0.104 (0.046) +0.208 (0.047) −0.271 (0.045) +0.041 (0.045) +0.312 (0.057) | 0.000 0.944 0.648 0.290 0.603 0.283 0.220 0.000 0.026 0.000 0.000 0.362 0.000 |
| ADCOMS | Time VShet−/ε4− v. VShet−/ε4+ VShet+/ε4− v. VShet−/ε4− VShet+/ε4+ v. VShet−/ε4− VShet+/ε4+ v. VShet−/ε4− VShet+/ε4+ v. VShet−/ε4+ VShet+/ε4+ v. VShet+/ε4− VShet−/ε4− v. VShet−/ε4+ x Time VShet+/ε4− v. VShet−/ε4− x Time VShet+/ε4+ v. VShet−/ε4− x Time VShet+/ε4− v. VShet−/ε4+ x Time VShet+/ε4+ v. VShet−/ε4+ x Time VShet+/ε4+ v. VShet+/ε4− x Time | +0.128 (0.007) −0.008 (0.032) −0.137 (0.060) +0.020 (0.046) −0.145 (0.058) +0.029 (0.042) +0.117 (0.066) −0.033 (0.006) −0.042 (0.009) +0.024 (0.009) −0.075 (0.008) +0.009 (0.008) +0.066 (0.010) | 0.000 0.791 0.023 0.662 0.012 0.500 0.080 0.000 0.000 0.005 0.000 0.243 0.000 |
| MMSE | CDR-SB | ADCOMS | |
|---|---|---|---|
| VShet−/ε4− | −1.067 (0.049) | +1.056 (0.028) | +0.082 (0.005) |
| VShet−/ε4+ | −1.423 (0.038) | +1.216 (0.022) | +0.112 (0.004) |
| VShet+/ε4− | −0.735 (0.081) | +0.903 (0.041) | +0.047 (0.004) |
| VShet+/ε4+ | −1.358 (0.087) | +1.312 (0.046) | +0.122 (0.008) |
| MMSE | CDR-SB | |||||
|---|---|---|---|---|---|---|
| Stratification | n | β (SE) | p | β (SE) | p | |
| Female | 310 | VShet+/ε4− | +0.039 (0.152) | 0.795 | +0.015 (0.070) | 0.834 |
| Male | 355 | VShet+/ε4− | +0.368 (0.109) | 0.001 | −0.196 (0.061) | 0.001 ** |
| Baseline age ≤ 76 yr. | 350 | VShet+/ε4− | −0.029 (0.175) | 0.867 | +0.047 (0.095) | 0.621 |
| Baseline age > 76 yr. | 315 | VShet+/ε4− | +0.520 (0.101) | 0.000 | −0.285 (0.055) | 0.000 ** |
| Education < 16 years | 231 | VShet+/ε4− | −0.025 (0.177) | 0.885 | +0.136 (0.087) | 0.117 |
| Education ≥ 16 years | 434 | VShet+/ε4− | +0.287 (0.089) | 0.001 | −0.104 (0.046) | 0.026 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.R.; Shao, Y.; Sadowski, M.J.; on behalf of the Alzheimer’s Disease Neuroimaging Initiative. Interaction between KLOTHO-VS Heterozygosity and APOE ε4 Allele Predicts Rate of Cognitive Decline in Late-Onset Alzheimer’s Disease. Genes 2023, 14, 917. https://doi.org/10.3390/genes14040917
Chen XR, Shao Y, Sadowski MJ, on behalf of the Alzheimer’s Disease Neuroimaging Initiative. Interaction between KLOTHO-VS Heterozygosity and APOE ε4 Allele Predicts Rate of Cognitive Decline in Late-Onset Alzheimer’s Disease. Genes. 2023; 14(4):917. https://doi.org/10.3390/genes14040917
Chicago/Turabian StyleChen, Xi Richard, Yongzhao Shao, Martin J. Sadowski, and on behalf of the Alzheimer’s Disease Neuroimaging Initiative. 2023. "Interaction between KLOTHO-VS Heterozygosity and APOE ε4 Allele Predicts Rate of Cognitive Decline in Late-Onset Alzheimer’s Disease" Genes 14, no. 4: 917. https://doi.org/10.3390/genes14040917
APA StyleChen, X. R., Shao, Y., Sadowski, M. J., & on behalf of the Alzheimer’s Disease Neuroimaging Initiative. (2023). Interaction between KLOTHO-VS Heterozygosity and APOE ε4 Allele Predicts Rate of Cognitive Decline in Late-Onset Alzheimer’s Disease. Genes, 14(4), 917. https://doi.org/10.3390/genes14040917