
A Multi-Tissue Gene Expression Atlas of Water Buffalo (Bubalus bubalis) Reveals Transcriptome Conservation between Buffalo and Cattle


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA-Seq Samples
2.2. Genome Similarity, Transcriptome Similarity and Homologous Gene Identification between Buffalo and Cattle
2.3. Sample Clustering
2.4. Detection of Tissue-Specific Genes (TSGs)
2.5. Detection of House-Keeping Genes (HKGs)
2.6. Detection of Differentially Expressed Genes (DEGs) between Species
3. Results
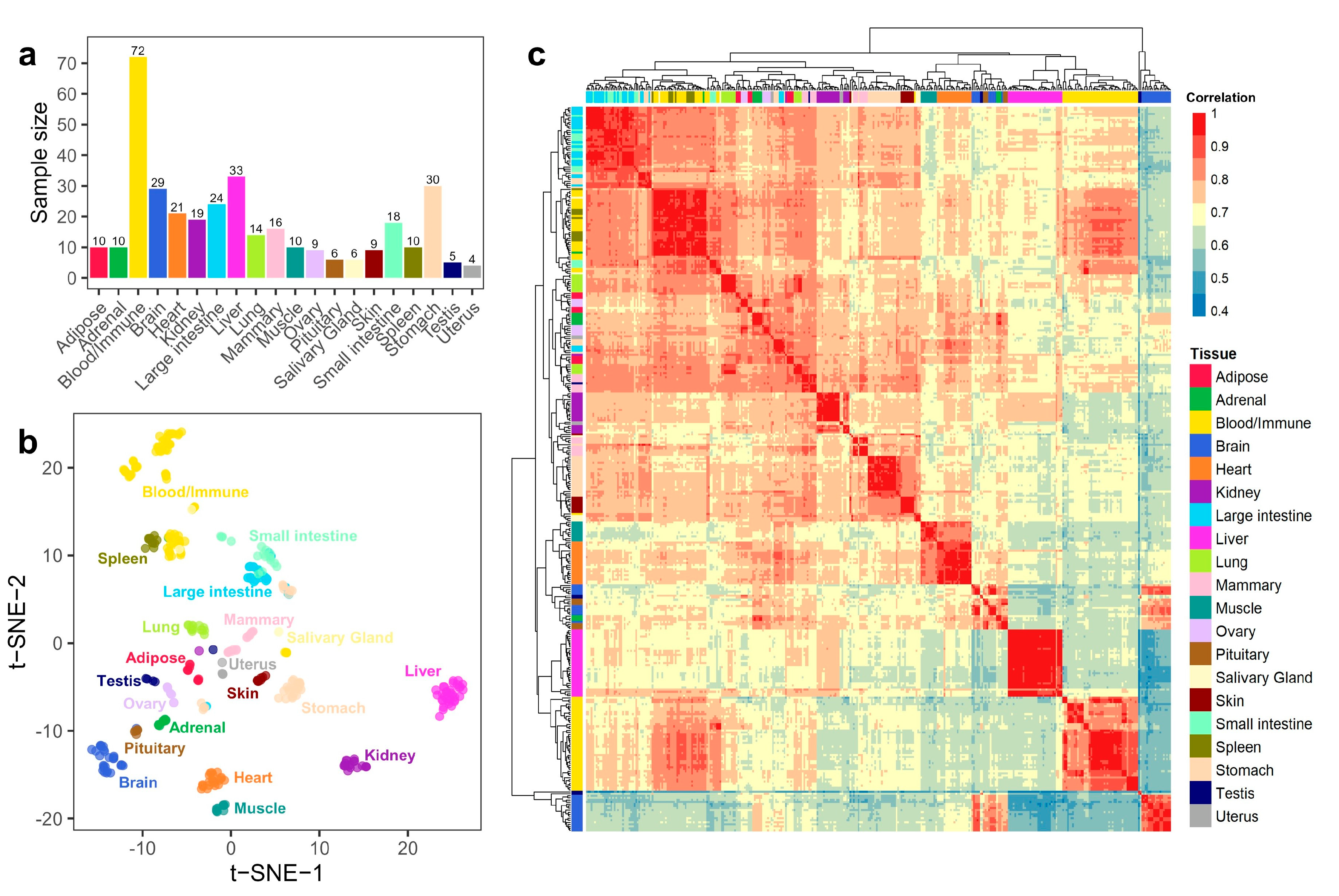
3.1. Summary of Gene Expression Profiles in Buffalo
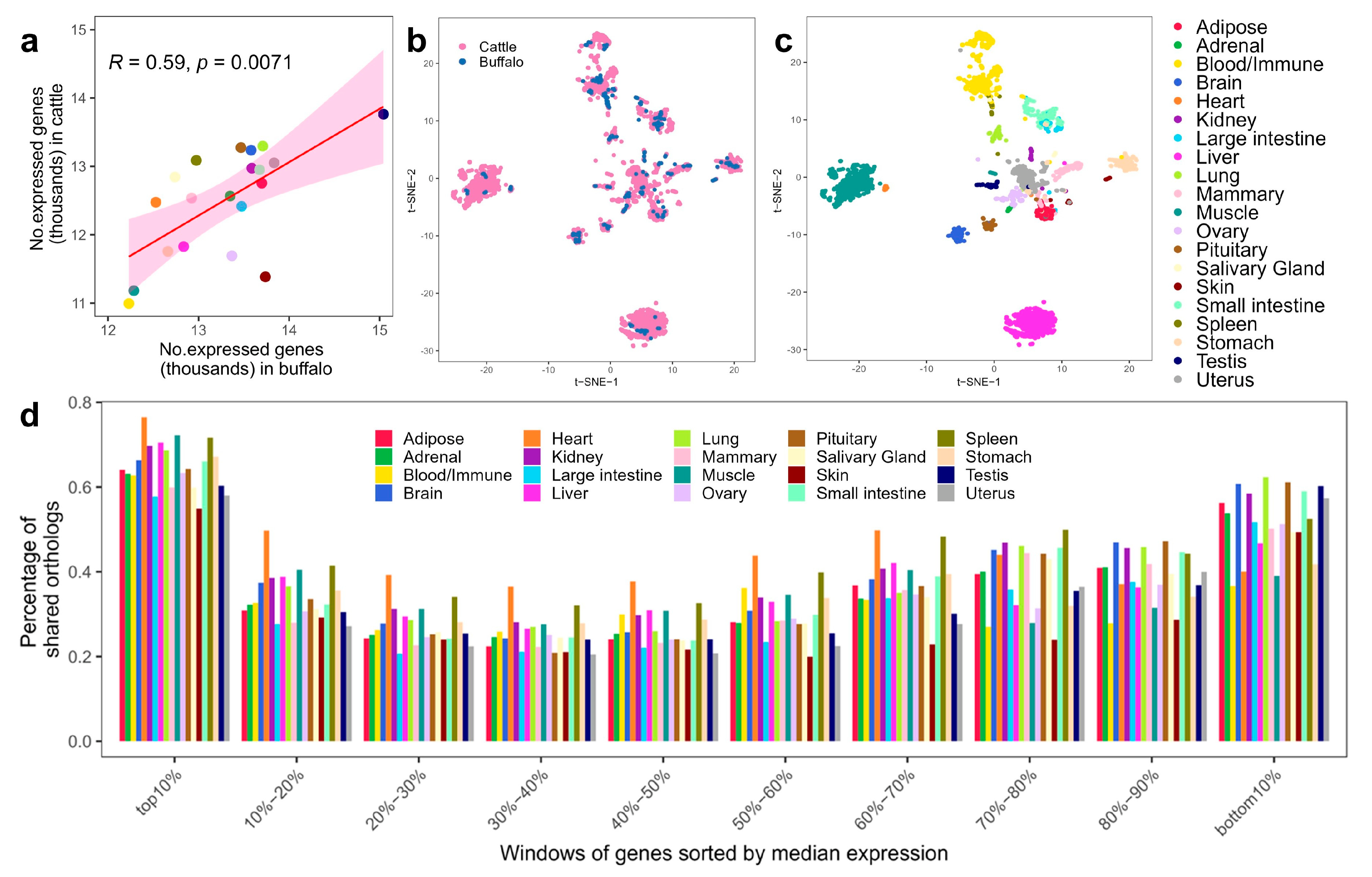
3.2. Sequence Similarity of Genome and Transcriptome between Buffalo and Cattle
3.3. Conservation of Global Gene Expression Patterns between Buffalo and Cattle
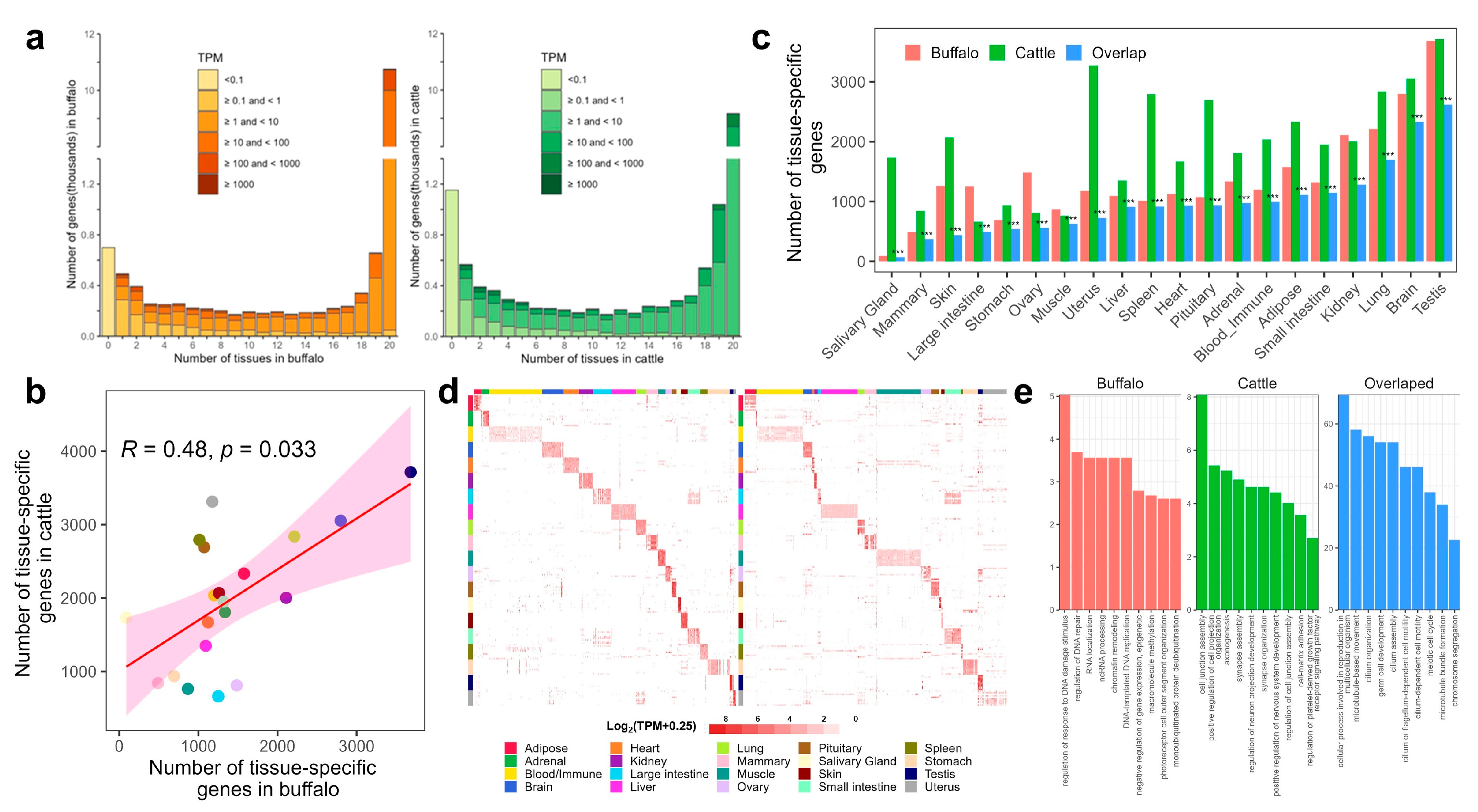
3.4. Detection and Comparison of Tissue-Specific Genes (TSGs)
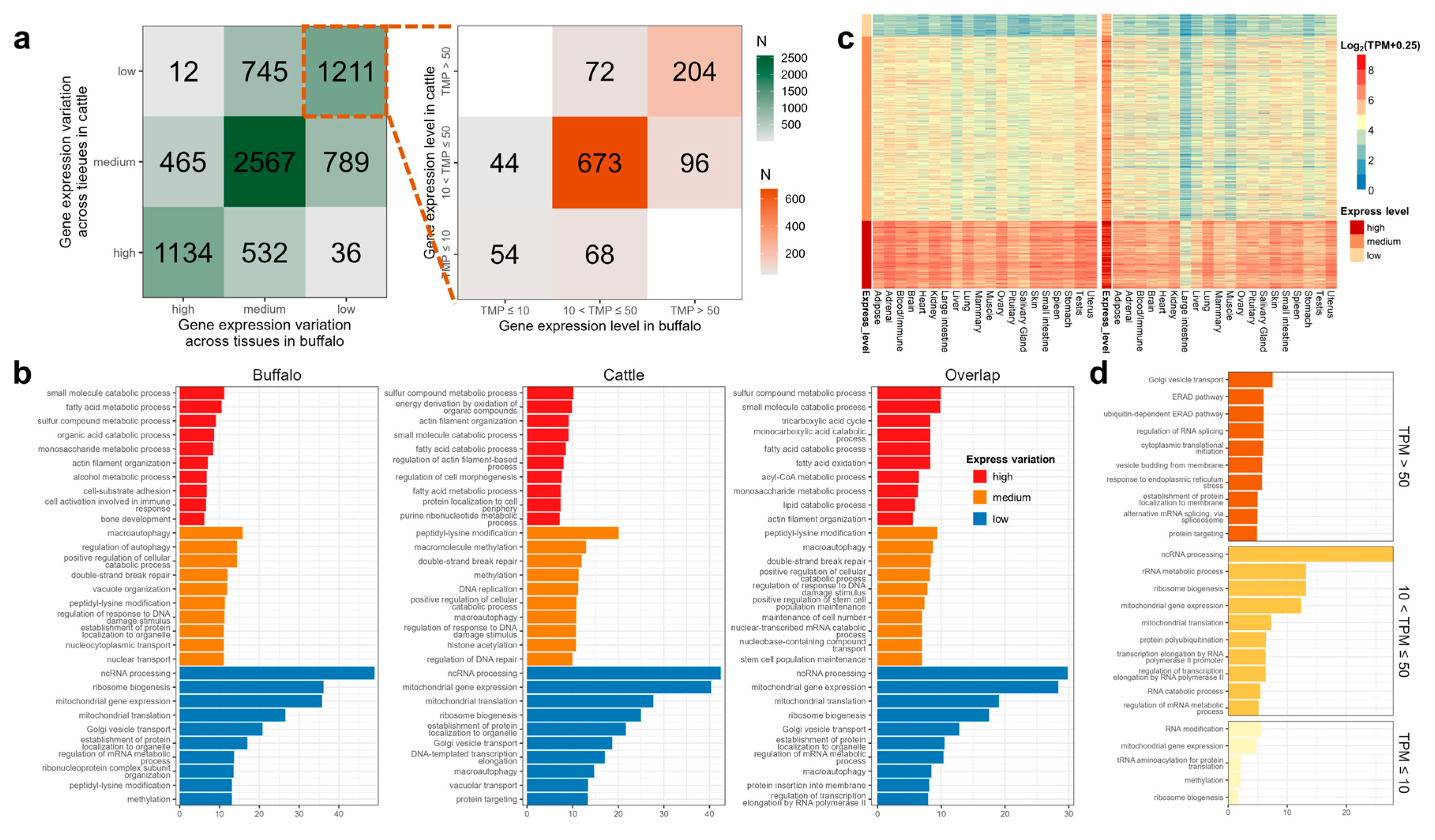
3.5. Detection and Comparison of House-Keeping Genes (HKGs)
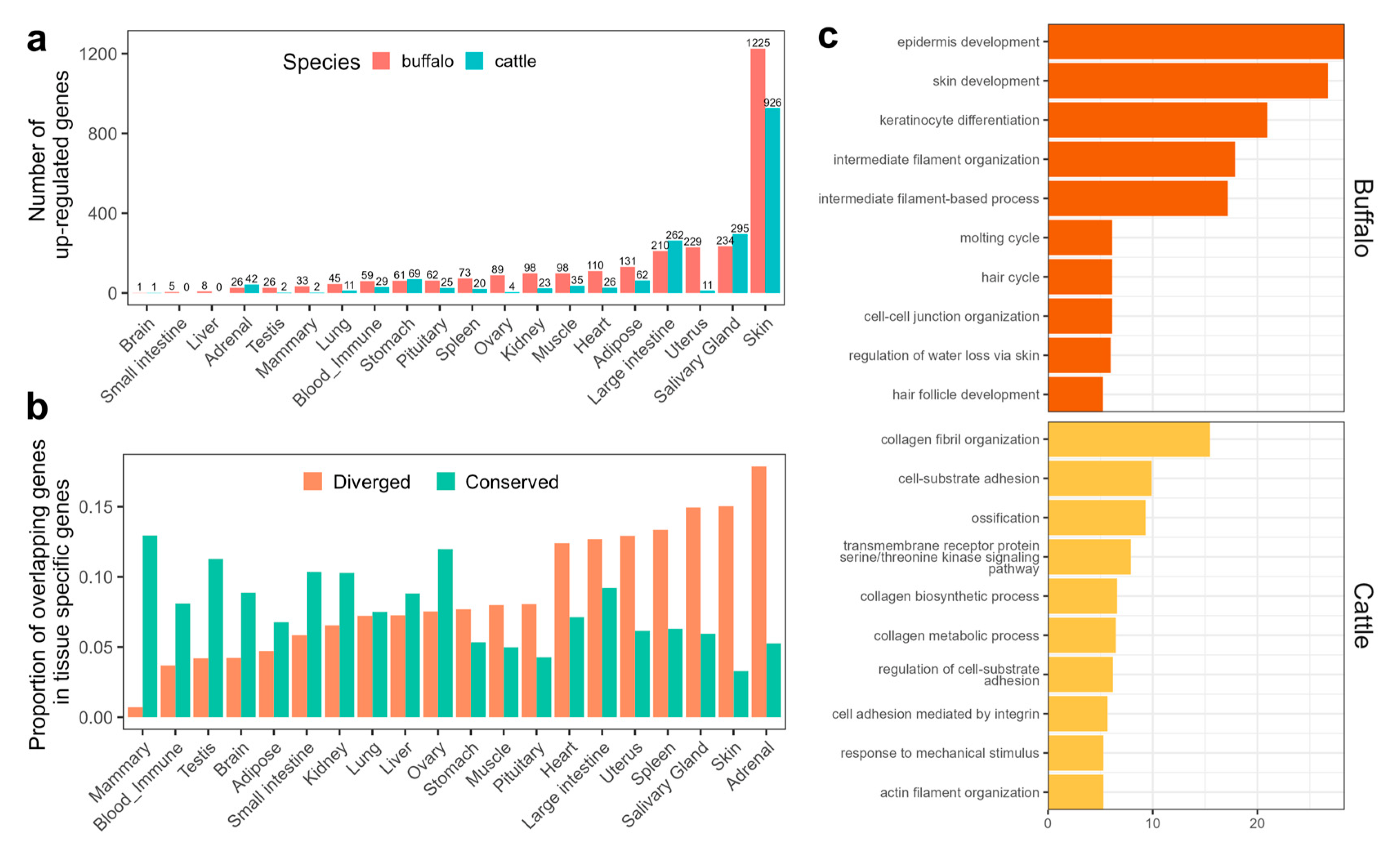
3.6. Detection of Differentially Expressed Genes (DEGs) between Species
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lizio, M.; Harshbarger, J.; Shimoji, H.; Severin, J.; Kasukawa, T.; Sahin, S.; Abugessaisa, I.; Fukuda, S.; Hori, F.; Ishikawa-Kato, S.; et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P.; Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Mele, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. Human genomics. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef]
- Stunnenberg, H.G.; International Human Epigenome, C.; Hirst, M. The International Human Epigenome Consortium: A Blueprint for Scientific Collaboration and Discovery. Cell 2016, 167, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gao, Y.; Canela-Xandri, O.; Wang, S.; Yu, Y.; Cai, W.; Li, B.; Xiang, R.; Chamberlain, A.J.; Pairo-Castineira, E.; et al. A multi-tissue atlas of regulatory variants in cattle. Nat. Genet. 2022, 54, 1438–1447. [Google Scholar] [CrossRef]
- Teng, J.; Gao, Y.; Yin, H.; Bai, Z.; Liu, S.; Zeng, H.; Bai, L.; Cai, Z.; Zhao, B.; Li, X.; et al. A compendium of genetic regulatory effects across pig tissues. bioRxiv 2022. [Google Scholar]
- Zhang, T.; Wang, T.; Niu, Q.; Xu, L.; Chen, Y.; Gao, X.; Gao, H.; Zhang, L.; Liu, G.E.; Li, J.; et al. Transcriptional atlas analysis from multiple tissues reveals the expression specificity patterns in beef cattle. BMC Biol. 2022, 20, 79. [Google Scholar] [CrossRef]
- Pan, X.; Cai, J.; Wang, Y.; Xu, D.; Jiang, Y.; Gong, W.; Tian, Y.; Shen, Q.; Zhang, Z.; Yuan, X.; et al. Expression Profile of Housekeeping Genes and Tissue-Specific Genes in Multiple Tissues of Pigs. Animals 2022, 12, 3539. [Google Scholar] [CrossRef]
- Cockrill, W.R. Evolution of domesticated Animals. In Water Buffalo; Mason, I.L., Ed.; Longman: London, UK, 1984; pp. 52–63. [Google Scholar]
- Zhang, Y.; Colli, L.; Barker, J.S.F. Asian water buffalo: Domestication, history and genetics. Anim. Genet. 2020, 51, 177–191. [Google Scholar] [CrossRef]
- Deb, G.K.; Nahar, T.N.; Duran, P.G.; Presicce, G.A. Safe and Sustainable Traditional Production: The Water Buffalo in Asia. Front. Environ. Sci. 2016, 4, 38. [Google Scholar] [CrossRef]
- Zicarelli, L. Buffalo milk: Its properties, dairy yield and mozzarella production. Vet. Res. Commun. 2004, 28 (Suppl. S1), 127–135. [Google Scholar] [CrossRef]
- Borghese, A.; Moioli, B. Buffalo: Mediterranean Region. In Encyclopedia of Dairy Sciences, 3rd ed.; McSweeney, P.L.H., McNamara, J.P., Eds.; Academic Press: Oxford, UK, 2016; pp. 845–849. [Google Scholar] [CrossRef]
- Iannuzzi, A.; Parma, P.; Iannuzzi, L. The Cytogenetics of the Water Buffalo: A Review. Animals 2021, 11, 3109. [Google Scholar] [CrossRef] [PubMed]
- Young, R.; Lefevre, L.; Bush, S.J.; Joshi, A.; Singh, S.H.; Jadhav, S.K.; Dhanikachalam, V.; Lisowski, Z.M.; Iamartino, D.; Summers, K.M.; et al. A Gene Expression Atlas of the Domestic Water Buffalo (Bubalus bubalis). Front. Genet. 2019, 10, 668. [Google Scholar] [CrossRef]
- Low, W.Y.; Tearle, R.; Bickhart, D.M.; Rosen, B.D.; Kingan, S.B.; Swale, T.; Thibaud-Nissen, F.; Murphy, T.D.; Young, R.; Lefevre, L.; et al. Chromosome-level assembly of the water buffalo genome surpasses human and goat genomes in sequence contiguity. Nat. Commun. 2019, 10, 260. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Jain, C.; Koren, S.; Dilthey, A.; Phillippy, A.M.; Aluru, S. A fast adaptive algorithm for computing whole-genome homology maps. Bioinformatics 2018, 34, i748–i756. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Kellis, M. Comparison of human and mouse tissues with focus on genes with no 1-to-1 homology. bioRxiv 2021. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e1821. [Google Scholar] [CrossRef]
- Yao, Y.; Liu, S.; Xia, C.; Gao, Y.; Pan, Z.; Canela-Xandri, O.; Khamseh, A.; Rawlik, K.; Wang, S.; Li, B.; et al. Comparative transcriptome in large-scale human and cattle populations. Genome Biol. 2022, 23, 176. [Google Scholar] [CrossRef] [PubMed]
- Van Der Maaten, L. Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res. 2014, 15, 3221–3245. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids. Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B-Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- de Jonge, H.J.; Fehrmann, R.S.; de Bont, E.S.; Hofstra, R.M.; Gerbens, F.; Kamps, W.A.; de Vries, E.G.; van der Zee, A.G.; te Meerman, G.J.; ter Elst, A. Evidence based selection of housekeeping genes. PLoS ONE 2007, 2, e8982007. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Lin, Y.; Nery, J.R.; Urich, M.A.; Breschi, A.; Davis, C.A.; Dobin, A.; Zaleski, C.; Beer, M.A.; Chapman, W.C.; et al. Comparison of the transcriptional landscapes between human and mouse tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 17224–17229. [Google Scholar] [CrossRef]
- Fang, L.; Cai, W.; Liu, S.; Canela-Xandri, O.; Gao, Y.; Jiang, J.; Rawlik, K.; Li, B.; Schroeder, S.G.; Rosen, B.D.; et al. Comprehensive analyses of 723 transcriptomes enhance genetic and biological interpretations for complex traits in cattle. Genome Res. 2020, 30, 790–801. [Google Scholar] [CrossRef]
- Djureinovic, D.; Fagerberg, L.; Hallstrom, B.; Danielsson, A.; Lindskog, C.; Uhlen, M.; Ponten, F. The human testis-specific proteome defined by transcriptomics and antibody-based profiling. Mol. Hum. Reprod. 2014, 20, 476–488. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Liu, S.; Zhao, Y.; Tan, X.; Aljohi, H.A.; Liu, W.; Hu, S. Identification and analysis of house-keeping and tissue-specific genes based on RNA-seq data sets across 15 mouse tissues. Gene 2016, 576, 560–570. [Google Scholar] [CrossRef] [PubMed]
- She, X.; Rohl, C.A.; Castle, J.C.; Kulkarni, A.V.; Johnson, J.M.; Chen, R. Definition, conservation and epigenetics of housekeeping and tissue-enriched genes. BMC Genom. 2009, 10, 269. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef] [PubMed]
- Jafari, H.; Lista, A.; Siekapen, M.M.; Ghaffari-Bohlouli, P.; Nie, L.; Alimoradi, H.; Shavandi, A. Fish Collagen: Extraction, Characterization, and Applications for Biomaterials Engineering. Polymers 2020, 12, 2230. [Google Scholar] [CrossRef]
- Matinong, A.M.E.; Chisti, Y.; Pickering, K.L.; Haverkamp, R.G. Collagen Extraction from Animal Skin. Biology 2022, 11, 905. [Google Scholar] [CrossRef]
- Hafez, E.S.E.; Badreldin, A.L.; Shafei, M.M. Skin structure of Egyptian buffaloes and cattle with particular reference to sweat glands. J. Agric. Sci. 2009, 46, 19–30. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Si, J.; Dai, D.; Li, K.; Fang, L.; Zhang, Y. A Multi-Tissue Gene Expression Atlas of Water Buffalo (Bubalus bubalis) Reveals Transcriptome Conservation between Buffalo and Cattle. Genes 2023, 14, 890. https://doi.org/10.3390/genes14040890
Si J, Dai D, Li K, Fang L, Zhang Y. A Multi-Tissue Gene Expression Atlas of Water Buffalo (Bubalus bubalis) Reveals Transcriptome Conservation between Buffalo and Cattle. Genes. 2023; 14(4):890. https://doi.org/10.3390/genes14040890
Chicago/Turabian StyleSi, Jingfang, Dongmei Dai, Kun Li, Lingzhao Fang, and Yi Zhang. 2023. "A Multi-Tissue Gene Expression Atlas of Water Buffalo (Bubalus bubalis) Reveals Transcriptome Conservation between Buffalo and Cattle" Genes 14, no. 4: 890. https://doi.org/10.3390/genes14040890
APA StyleSi, J., Dai, D., Li, K., Fang, L., & Zhang, Y. (2023). A Multi-Tissue Gene Expression Atlas of Water Buffalo (Bubalus bubalis) Reveals Transcriptome Conservation between Buffalo and Cattle. Genes, 14(4), 890. https://doi.org/10.3390/genes14040890