
Leishmania infantum (JPCM5) Transcriptome, Gene Models and Resources for an Active Curation of Gene Annotations

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Leishmania Parasites
2.2. RNA Isolation
2.3. Illumina RNA-Seq and Data Processing
2.4. Iso-Seq Analyses
2.5. PCR Assays
2.6. Gene-Model Annotation
2.7. Determination of RNA Levels and Differential Expression from RNA-Seq Data
2.8. Data Availability
3. Results and Discussion
3.1. Delineation of the Poly-A+ Transcriptome for the L. infantum (JPCM5 Strain) Promastigote Stage Based on Illumina RNA-Seq Methodology
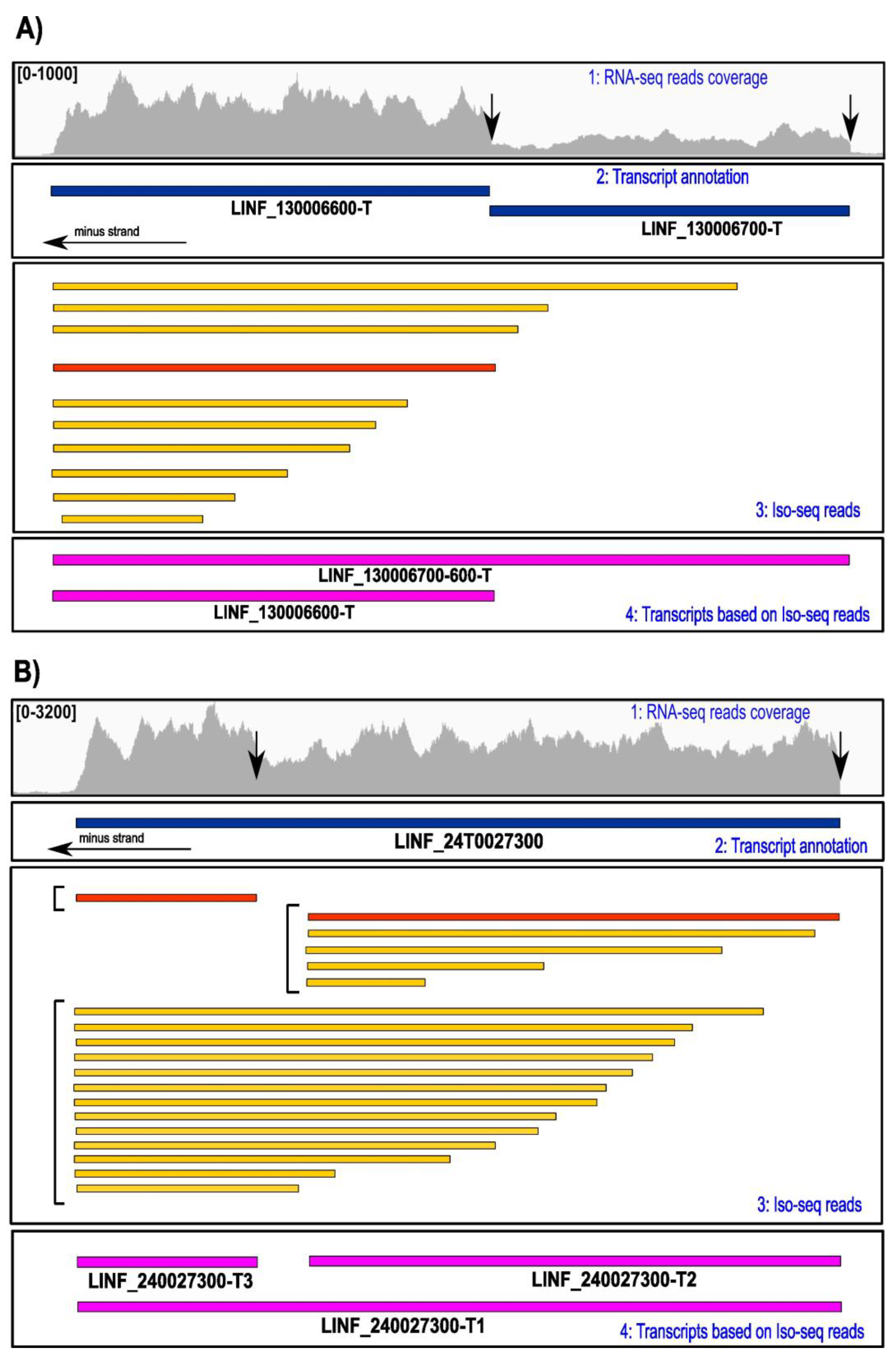
3.2. Refinement of the Transcriptome by the Iso-Seq Methodology
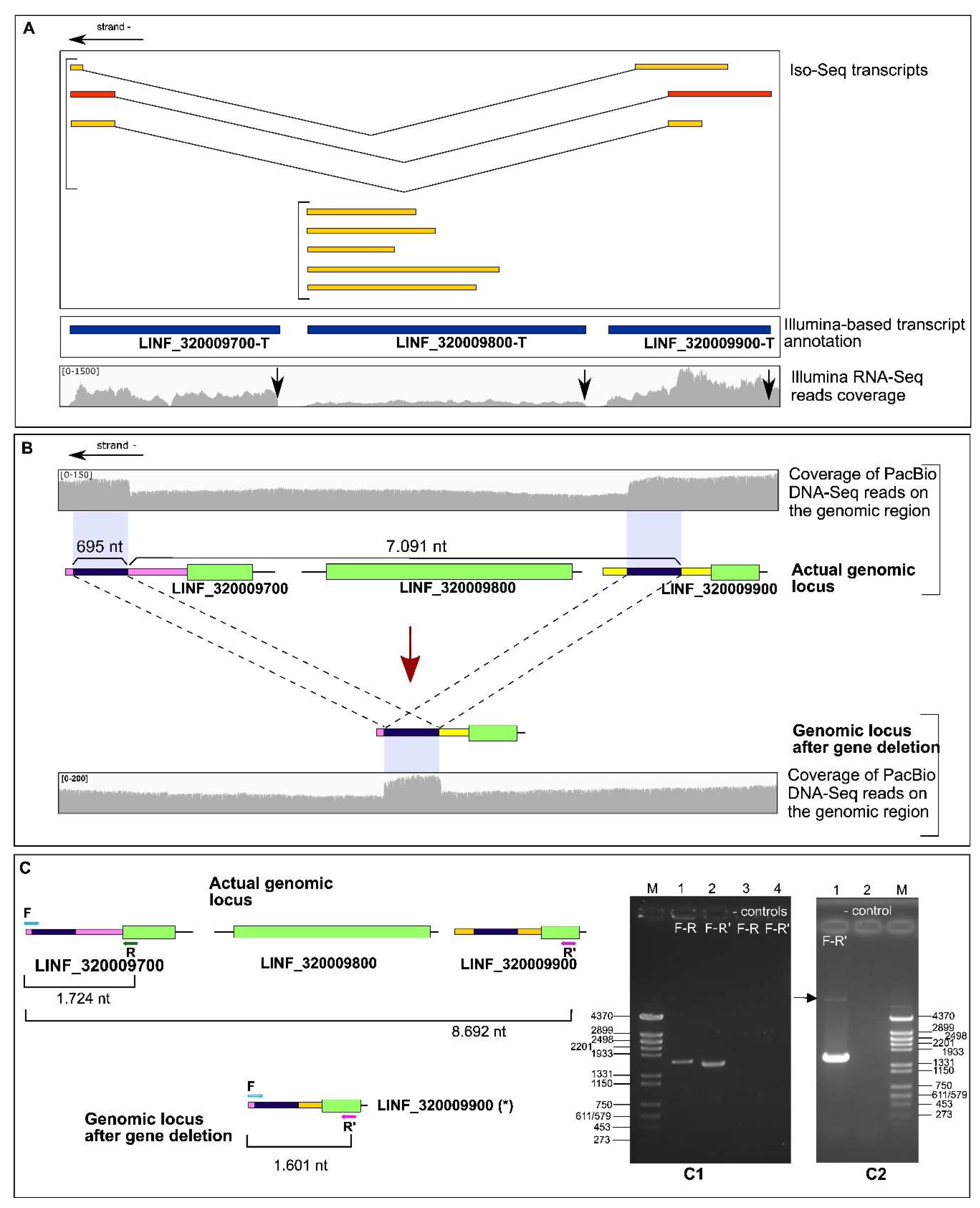
3.3. Identification of an Allelic Deletion (Hemizygosity) Affecting Genes LINF_320009700, LINF_320009800 and LINF_320009900
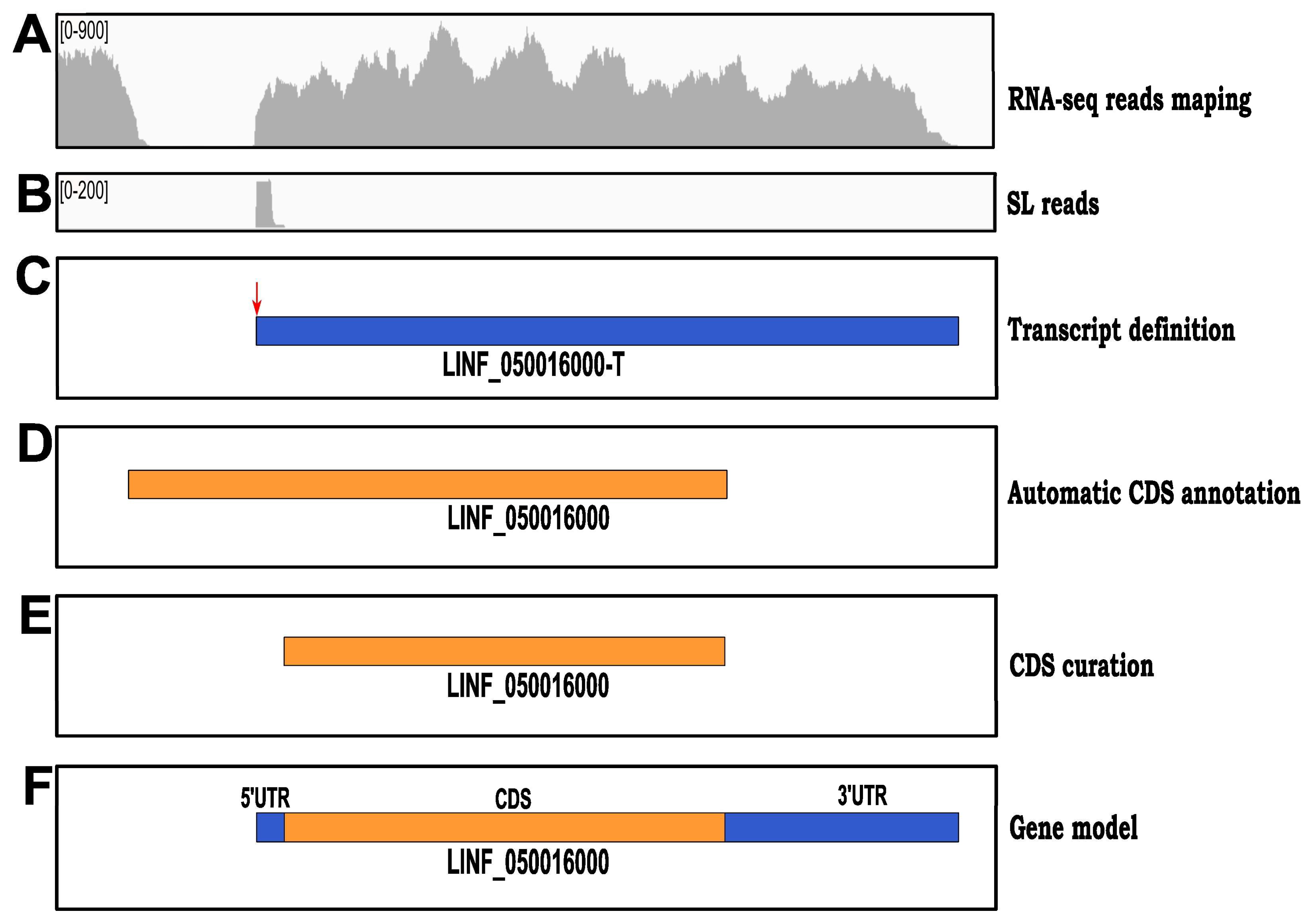
3.4. Setting Up Gene Models
3.5. Relative Expression Levels of Transcripts in L. infantum Promastigotes
3.6. Species-Specific Differences in the Steady-State Transcript Abundance
3.7. Active Curation of Gene Models and Annotations Based on Wikidata and Mendeley Data Platforms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alvar, J.; Velez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef]
- Volpedo, G.; Huston, R.H.; Holcomb, E.A.; Pacheco-Fernandez, T.; Gannavaram, S.; Bhattacharya, P.; Nakhasi, H.L.; Satoskar, A.R. From infection to vaccination: Reviewing the global burden, history of vaccine development, and recurring challenges in global leishmaniasis protection. Expert Rev. Vaccines 2021, 20, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.S.; Seeger, K.; Harris, D.; Murphy, L.; Ruiz, J.C.; Quail, M.A.; Peters, N.; Adlem, E.; Tivey, A.; Aslett, M.; et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 2007, 39, 839–847. [Google Scholar] [CrossRef]
- Rochette, A.; Raymond, F.; Corbeil, J.; Ouellette, M.; Papadopoulou, B. Whole-genome comparative RNA expression profiling of axenic and intracellular amastigote forms of Leishmania infantum. Mol. Biochem. Parasitol. 2009, 165, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, D.; Smith, D.; Opperdoes, F.; Stern, S.; Olafson, R.W.; Zilberstein, D. Retooling Leishmania metabolism: From sand fly gut to human macrophage. Faseb J. 2008, 22, 590–602. [Google Scholar] [CrossRef]
- Dikhit, M.R.; Kumar, A.; Das, S.; Dehury, B.; Rout, A.K.; Jamal, F.; Sahoo, G.C.; Topno, R.K.; Pandey, K.; Das, V.N.R.; et al. Identification of Potential MHC Class-II-Restricted Epitopes Derived from Leishmania donovani Antigens by Reverse Vaccinology and Evaluation of Their CD4+ T-Cell Responsiveness against Visceral Leishmaniasis. Front. Immunol. 2017, 8, 1763. [Google Scholar] [CrossRef]
- Gonzalez-de la Fuente, S.; Peiro-Pastor, R.; Rastrojo, A.; Moreno, J.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Resequencing of the Leishmania infantum (strain JPCM5) genome and de novo assembly into 36 contigs. Sci. Rep. 2017, 7, 18050. [Google Scholar] [CrossRef]
- Aslett, M.; Aurrecoechea, C.; Berriman, M.; Brestelli, J.; Brunk, B.P.; Carrington, M.; Depledge, D.P.; Fischer, S.; Gajria, B.; Gao, X.; et al. TriTrypDB: A functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010, 38, D457–D462. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Clayton, C.; Shapira, M. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol. Biochem. Parasitol. 2007, 156, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Moya, S.M.; Estevez, A.M. Posttranscriptional control and the role of RNA-binding proteins in gene regulation in trypanosomatid protozoan parasites. Wiley Interdiscip. Rev. RNA 2010, 1, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Requena, J.M. Lights and shadows on gene organization and regulation of gene expression in Leishmania. Front. Biosci. 2011, 16, 2069–2085. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Calvillo, S.; Vizuet-de-Rueda, J.C.; Florencio-Martinez, L.E.; Manning-Cela, R.G.; Figueroa-Angulo, E.E. Gene expression in trypanosomatid parasites. J. Biomed. Biotechnol. 2010, 2010, 525241. [Google Scholar] [CrossRef] [PubMed]
- LeBowitz, J.H.; Smith, H.Q.; Rusche, L.; Beverley, S.M. Coupling of poly(A) site selection and trans-splicing in Leishmania. Genes Dev. 1993, 7, 996–1007. [Google Scholar] [CrossRef]
- Liang, X.H.; Haritan, A.; Uliel, S.; Michaeli, S. trans and cis splicing in trypanosomatids: Mechanism, factors, and regulation. Eukaryot. Cell 2003, 2, 830–840. [Google Scholar] [CrossRef]
- Mair, G.; Shi, H.; Li, H.; Djikeng, A.; Aviles, H.O.; Bishop, J.R.; Falcone, F.H.; Gavrilescu, C.; Montgomery, J.L.; Santori, M.I.; et al. A new twist in trypanosome RNA metabolism: Cis-splicing of pre-mRNA. RNA 2000, 6, 163–169. [Google Scholar] [CrossRef]
- Jae, N.; Wang, P.; Gu, T.; Huhn, M.; Palfi, Z.; Urlaub, H.; Bindereif, A. Essential role of a trypanosome U4-specific Sm core protein in small nuclear ribonucleoprotein assembly and splicing. Eukaryot. Cell 2010, 9, 379–386. [Google Scholar] [CrossRef]
- Camacho, E.; González-de la Fuente, S.; Rastrojo, A.; Peiró-Pastor, R.; Solana, J.C.; Tabera, L.; Gamarro, F.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Complete assembly of the Leishmania donovani (HU3 strain) genome and transcriptome annotation. Sci. Rep. 2019, 9, 6127. [Google Scholar] [CrossRef] [PubMed]
- Kramer, S.; Carrington, M. Trans-acting proteins regulating mRNA maturation, stability and translation in trypanosomatids. Trends Parasitol. 2011, 27, 23–30. [Google Scholar] [CrossRef]
- Siegel, T.N.; Gunasekera, K.; Cross, G.A.M.; Ochsenreiter, T. Gene expression in Trypanosoma brucei: Lessons from high-throughput RNA sequencing. Trends Parasitol. 2011, 27, 434–441. [Google Scholar] [CrossRef]
- Rastrojo, A.; Carrasco-Ramiro, F.; Martín, D.; Crespillo, A.; Reguera, R.M.; Aguado, B.; Requena, J.M. The transcriptome of Leishmania major in the axenic promastigote stage: Transcript annotation and relative expression levels by RNA-seq. BMC Genom. 2013, 14, 223. [Google Scholar] [CrossRef] [PubMed]
- Fiebig, M.; Kelly, S.; Gluenz, E. Comparative Life Cycle Transcriptomics Revises Leishmania mexicana Genome Annotation and Links a Chromosome Duplication with Parasitism of Vertebrates. PLoS Pathog. 2015, 11, e1005186. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.; Nieto, J.; Masina, S.; Cañavate, C.; Cruz, I.; Chicharro, C.; Carrillo, E.; Napp, S.; Reymond, C.; Kaye, P.M.; et al. Immunization with H1, HASPB1 and MML Leishmania proteins in a vaccine trial against experimental canine leishmaniasis. Vaccine 2007, 25, 5290–5300. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef]
- Salas, M. 40 years with bacteriophage ø29. Annu. Rev. Microbiol. 2007, 61, 1–22. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhu, J.; Tong, T.; Wang, J.; Lin, B.; Zhang, J. A statistical normalization method and differential expression analysis for RNA-seq data between different species. BMC Bioinform. 2019, 20, 163. [Google Scholar] [CrossRef]
- Fiebig, M.; Gluenz, E.; Carrington, M.; Kelly, S. SLaP mapper: A webserver for identifying and quantifying spliced-leader addition and polyadenylation site usage in kinetoplastid genomes. Mol. Biochem. Parasitol. 2014, 196, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Kolev, N.G.; Franklin, J.B.; Carmi, S.; Shi, H.; Michaeli, S.; Tschudi, C. The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog. 2010, 6, e1001090. [Google Scholar] [CrossRef] [PubMed]
- Camacho, E.; González-de la Fuente, S.; Solana, J.C.; Rastrojo, A.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Gene annotation and transcriptome delineation on a de novo genome assembly for the reference Leishmania major Friedlin strain. Genes 2021, 12, 1359. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.C.; Ephrussi, A. mRNA localization: Gene expression in the spatial dimension. Cell 2009, 136, 719–730. [Google Scholar] [CrossRef]
- Mayr, C. Regulation by 3’-Untranslated Regions. Annu. Rev. Genet. 2017, 51, 171–194. [Google Scholar] [CrossRef]
- Steinbiss, S.; Silva-Franco, F.; Brunk, B.; Foth, B.; Hertz-Fowler, C.; Berriman, M.; Otto, T.D. Companion: A web server for annotation and analysis of parasite genomes. Nucleic Acids Res. 2016, 44, W29–W34. [Google Scholar] [CrossRef] [PubMed]
- Rascher, C.; Pahl, A.; Pecht, A.; Brune, K.; Solbach, W.; Bang, H. Leishmania major parasites express cyclophilin isoforms with an unusual interaction with calcineurin. Biochem. J. 1998, 334, 659–667. [Google Scholar] [CrossRef]
- Paterou, A.; Walrad, P.; Craddy, P.; Fenn, K.; Matthews, K. Identification and stage-specific association with the translational apparatus of TbZFP3, a CCCH protein that promotes trypanosome life-cycle development. J. Biol. Chem. 2006, 281, 39002–39013. [Google Scholar] [CrossRef]
- da Costa, K.S.; Galucio, J.M.P.; Leonardo, E.S.; Cardoso, G.; Leal, E.; Conde, G.; Lameira, J. Structural and evolutionary analysis of Leishmania Alba proteins. Mol. Biochem. Parasitol. 2017, 217, 23–31. [Google Scholar] [CrossRef]
- Arastu-Kapur, S.; Ford, E.; Ullman, B.; Carter, N.S. Functional analysis of an inosine-guanosine transporter from Leishmania donovani: The role of conserved residues, aspartate 389 and arginine 393. J. Biol. Chem. 2003, 278, 33327–33333. [Google Scholar] [CrossRef]
- Maslov, D.A.; Opperdoes, F.R.; Kostygov, A.Y.; Hashimi, H.; Lukes, J.; Yurchenko, V. Recent advances in trypanosomatid research: Genome organization, expression, metabolism, taxonomy and evolution. Parasitology 2019, 146, 1–27. [Google Scholar] [CrossRef]
- Jardim, A.; Funk, V.; Caprioli, R.M.; Olafson, R.W. Isolation and structural characterization of the Leishmania donovani kinetoplastid membrane protein-11, a major immunoreactive membrane glycoprotein. Biochem. J. 1995, 305, 307–313. [Google Scholar] [CrossRef]
- Teixeira, F.; Castro, H.; Cruz, T.; Tse, E.; Koldewey, P.; Southworth, D.R.; Tomas, A.M.; Jakob, U. Mitochondrial peroxiredoxin functions as crucial chaperone reservoir in Leishmania infantum. Proc. Natl. Acad. Sci. USA 2015, 112, E616–E624. [Google Scholar] [CrossRef] [PubMed]
- Folgueira, C.; Quijada, L.; Soto, M.; Abanades, D.R.; Alonso, C.; Requena, J.M. The translational efficiencies of the two Leishmania infantum HSP70 mRNAs, differing in their 3′-untranslated regions, are affected by shifts in the temperature of growth through different mechanisms. J. Biol. Chem. 2005, 280, 35172–35183. [Google Scholar] [CrossRef] [PubMed]
- Kaye, P.M.; Cruz, I.; Picado, A.; Van Bocxlaer, K.; Croft, S.L. Leishmaniasis immunopathology—Impact on design and use of vaccines, diagnostics and drugs. Semin. Immunopathol. 2020, 42, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.B.; Hilley, J.D.; Dickens, N.J.; Wilkes, J.; Bates, P.A.; Depledge, D.P.; Harris, D.; Her, Y.; Herzyk, P.; Imamura, H.; et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011, 21, 2129–2142. [Google Scholar] [CrossRef] [PubMed]
- Mugo, E.; Clayton, C. Expression of the RNA-binding protein RBP10 promotes the bloodstream-form differentiation state in Trypanosoma brucei. PLoS Pathog. 2017, 13, e1006560. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, C.; Zander, D.; Fleischer, S.; Schilhabel, M.; Kroener, M.; Platzer, M.; Clos, J. A Leishmania donovani gene that confers accelerated recovery from stationary phase growth arrest. Int. J. Parasitol. 2004, 34, 803–811. [Google Scholar] [CrossRef]
- Amos, B.; Aurrecoechea, C.; Barba, M.; Barreto, A.; Basenko, E.Y.; Bażant, W.; Belnap, R.; Blevins, A.S.; Böhme, U.; Brestelli, J.; et al. VEuPathDB: The eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 2022, 50, D898–D911. [Google Scholar] [CrossRef]
- Sanchiz, Á.; Morato, E.; Rastrojo, A.; Camacho, E.; González-de la Fuente, S.; Marina, A.; Aguado, B.; Requena, J.M. The Experimental Proteome of Leishmania infantum Promastigote and Its Usefulness for Improving Gene Annotations. Genes 2020, 11, 1036. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Transcript ID | TPM (SD) * | Encoded Protein |
|---|---|---|
| LINF_270018300-T | 3059.1 (237.5) | putative histone H1 |
| LINF_060005000-T | 3045.2 (381.5) | histone H4 |
| LINF_130010600-T | 2872.2 (67.2) | ribosomal protein S12|eS12 |
| LINF_250014900-T | 2691.3 (60.7) | cyclophilin A|Cyp19 |
| LINF_190005200-T | 2456.7 (220.9) | histone H2B |
| LINF_350007500-T | 2398.6 (210.0) | ribosomal protein L30|eL30 |
| LINF_270006200-T | 2379.3 (68.1) | ZFP3 |
| LINF_130009400-T | 2362.5 (105.1) | Alba1 |
| LINF_270018800-T | 2356.2 (187.1) | putative histone H1 |
| LINF_290024100-T | 2262.3 (158.0) | histone H2A |
| LINF_210028500-T | 2160.8 (86.3) | β tubulin |
| LINF_100016800-T | 2086.0 (51.6) | histone H3 |
| LINF_090020900-T | 2082.2 (147.6) | histone H2B |
| LINF_260013700-T | 1850.5 (41.8) | ribosomal protein S16|uS9 |
| LINF_360026000-T | 1846.1 (131.0) | inosine-guanosine transporter|NT2 |
| LINF_100017000-T | 1795.0 (62.1) | histone H3 |
| LINF_260028900-T | 1786.9 (52.6) | ribosomal protein L35|uL29 |
| LINF_130017300-T | 1772.4 (55.9) | ribosomal protein S4|eS4 |
| LINF_300039100-T | 1753.7 (32.4) | ribosomal protein L9|uL6 |
| LINF_200021000-T | 1738.7 (61.0) | ribosomal protein S11|uS17 |
| LINF_260013800-T | 1708.7 (47.5) | ribosomal protein S16|uS9 |
| LINF_230018500-T | 1627.6 (268.5) | protein of unknown function-conserved |
| LINF_130022200-T | 1612.9 (120.3) | ribosomal protein L44|eL42 |
| LINF_260028800-T | 1611.7 (37.9) | ribosomal protein L35|uL29 |
| LINF_190005300-T | 1588.4 (79.4) | histone H2B |
| LINF_290023900-T | 1584.2 (11.2) | histone H2A |
| LINF_130017200-T | 1582.4 (48.0) | ribosomal protein S4|eS4 |
| LINF_030007300-T | 1578.7 (110.0) | ribosomal protein L38|eL38 |
| LINF_130008400-T | 1559.2 (65.8) | α tubulin |
| LINF_350043400-T | 1557.6 (56.9) | ribosomal protein L23|uL14 |
| LINF_290024000-T | 1557.3 (32.8) | histone H2A |
| LINF_020005100-T | 1554.9 (90.5) | histone H4 |
| LINF_130008800-T | 1551.6 (57.8) | α tubulin |
| LINF_350027400-T | 1538.8 (109.4) | kinetoplastid membrane protein-11 |
| LINF_240028300-T | 1535.0 (65.4) | ribosomal protein L12|uL11 |
| LINF_350010700-T | 1531.8 (49.9) | ribosomal protein L18a|eL20 |
| LINF_230005400-T | 1528.6 (150.7) | peroxidoxin mTXNPx |
| LINF_190005400-T | 1523.2 (50.3) | 40S ribosomal protein S2|uS5 |
| LINF_280035000-T | 1522.6 (156.0) | heat shock protein 70|HSP70-type II |
| LINF_320009400-T | 1520.9 (66.7) | ribosomal protein L17|uL22 |
| L. infantum Gene | L. major Gene | Ratio a | Gene Product |
|---|---|---|---|
| LINF_350031300 | LMJFC_350034400 | 16.56 | hypothetical protein-conserved |
| LINF_230013000 | LMJFC_230013700 | 16.00 | PAP2 superfamily |
| LINF_360079600 | LMJFC_360088700 | 12.29 | FCP1 domain-containing protein |
| LINF_300027400 | LMJFC_300032300 | 10.85 | Zinc finger (C3H1-type) domain-containing protein |
| LINF_220005200 | LMJFC_220005700 | 9.45 | hypothetical protein-conserved |
| LINF_350026500 | LMJFC_350028900 | 9.41 | protein of unknown function-conserved |
| LINF_010013300 | LMJFC_010013800 | 9.10 | potassium channel subunit-like protein |
| LINF_230009700 | LMJFC_230010800 | 7.06 | permease-like protein |
| LINF_230015000 | LMJFC_230016400 | 6.72 | RNA-binding protein|RBP10 |
| LINF_020009800 | LMJFC_020010200 | 6.35 | voltage-dependent anion-selective channel |
| LINF_290021400 | LMJFC_290023300 | 5.41 | Nodulin-like |
| LINF_290020300 | LMJFC_290022000 | 5.19 | RNA binding protein |
| LINF_120010100 | LMJFC_120011300 | 4.97 | GF1 |
| LINF_340047300 | LMJFC_340050800 | 4.74 | hypothetical protein-conserved |
| LINF_290019300 | LMJFC_290021100 | 4.59 | RNA-binding protein |
| LINF_260019950 | LMJFC_260021700 | 4.55 | protein of unknown function-conserved |
| LINF_310012900 | LMJFC_310013800 | 4.48 | protein of unknown function-conserved |
| LINF_360076300 | LMJFC_360084700 | 4.45 | tartrate-sensitive acid phosphatase |
| LINF_050016400 | LMJFC_050017700 | 4.36 | RNA-binding protein |
| LINF_350028500 | LMJFC_350030900 | 4.25 | protein kinase |
| LINF_090020400 | LMJFC_090020100 | 0.18 | PPPDE peptidase domain-containing protein |
| LINF_350036100 | LMJFC_350040300 | 0.17 | glycerol kinase-glycosomal |
| LINF_310005000 | LMJFC_310005200 | 0.15 | 5-methyltetrahydropteroyltriglutamate-homocysteine |
| S-methyltransferase | |||
| LINF_330008100 | LMJFC_330008500 | 0.15 | hypothetical protein-conserved |
| LINF_120013500 | LMJFC_120015000 | 0.15 | surface antigen protein 2 |
| LINF_060018800 | LMJFC_060019700 | 0.14 | pteridine transporter |
| LINF_290007800 | LMJFC_290008500 | 0.14 | D-lactate dehydrogenase-like protein |
| LINF_330038500 | LMJFC_330043100 | 0.14 | hypothetical protein-conserved |
| LINF_020005000 | LMJFC_020005200 | 0.13 | Side chain galactosyltransferase (SCG3) |
| LINF_310008800 | LMJFC_310009100 | 0.05 | amino acid transporter|AAT1.3 |
| LINF_220009700 | LMJFC_220010200 | 0.04 | hypothetical protein-conserved |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camacho, E.; González-de la Fuente, S.; Solana, J.C.; Tabera, L.; Carrasco-Ramiro, F.; Aguado, B.; Requena, J.M. Leishmania infantum (JPCM5) Transcriptome, Gene Models and Resources for an Active Curation of Gene Annotations. Genes 2023, 14, 866. https://doi.org/10.3390/genes14040866
Camacho E, González-de la Fuente S, Solana JC, Tabera L, Carrasco-Ramiro F, Aguado B, Requena JM. Leishmania infantum (JPCM5) Transcriptome, Gene Models and Resources for an Active Curation of Gene Annotations. Genes. 2023; 14(4):866. https://doi.org/10.3390/genes14040866
Chicago/Turabian StyleCamacho, Esther, Sandra González-de la Fuente, Jose Carlos Solana, Laura Tabera, Fernando Carrasco-Ramiro, Begoña Aguado, and Jose M. Requena. 2023. "Leishmania infantum (JPCM5) Transcriptome, Gene Models and Resources for an Active Curation of Gene Annotations" Genes 14, no. 4: 866. https://doi.org/10.3390/genes14040866
APA StyleCamacho, E., González-de la Fuente, S., Solana, J. C., Tabera, L., Carrasco-Ramiro, F., Aguado, B., & Requena, J. M. (2023). Leishmania infantum (JPCM5) Transcriptome, Gene Models and Resources for an Active Curation of Gene Annotations. Genes, 14(4), 866. https://doi.org/10.3390/genes14040866