Abstract
Reproductive traits have a key impact on production efficiency in the pig industry. It is necessary to identify the genetic structure of potential genes that influence reproductive traits. In this study, a genome-wide association study (GWAS) based on chip and imputed data of five reproductive traits, namely, total number born (TNB), number born alive (NBA), litter birth weight (LBW), gestation length (GL), and number of weaned (NW), was performed in Yorkshire pigs. In total, 272 of 2844 pigs with reproductive records were genotyped using KPS Porcine Breeding SNP Chips, and then chip data were imputed to sequencing data using two online software programs: the Pig Haplotype Reference Panel (PHARP v2) and Swine Imputation Server (SWIM 1.0). After quality control, we performed GWAS based on chip data and the two different imputation databases by using fixed and random model circulating probability unification (FarmCPU) models. We discovered 71 genome-wide significant SNPs and 25 potential candidate genes (e.g., SMAD4, RPS6KA2, CAMK2A, NDST1, and ADCY5). Functional enrichment analysis revealed that these genes are mainly enriched in the calcium signaling pathway, ovarian steroidogenesis, and GnRH signaling pathways. In conclusion, our results help to clarify the genetic basis of porcine reproductive traits and provide molecular markers for genomic selection in pig breeding.
1. Introduction
The reproductive performance of pigs plays a key role in the pig industry. Improving the reproductive performance of sows can lead to higher economic benefits for pig farms. However, reproductive traits are low-heritability traits, and their genetic structure is much more complex [1]. Therefore, it is difficult to improve these traits more rapidly using traditional breeding methods. With the development of molecular breeding technology, marker-assisted selection (MAS) and genomic selection (GS) have become effective ways to improve pig breeding efficiency [2].
In recent years, to complete genomic screening for trait-associated variants, genome-wide association studies (GWASs) have been widely applied to find quantitative trait loci (QTL) in economic traits [3]. Thus far, 35,384 QTLs have been identified in pigs according to pigQTLdb, of which 3315 QTLs are associated with reproduction (https://www.animalgenome.org/cgi-bin/QTLdb/SS/summary, 25 April 2022). In pigs, GWAS has identified numerous SNPs significantly associated with growth traits [4,5], meat quality [6,7], feed efficiency [8,9], semen traits [10,11], coat color [12,13], genetic defects [14,15], disease susceptibility [16,17], and microbial phenotypes [18]. However, most of them were genotyped based on SNP microarrays, and the density of markers is a key factor affecting GWAS efficiency [19]. With the development of sequencing technology and its increasingly low cost, many researchers have used sequencing or resequencing to perform relevant studies [20,21,22]. However, the sequencing or resequencing of large population samples is too costly and remains an inefficient strategy. Genotype imputation is an effective strategy in GWAS [23], which has been widely used in human genetics research, such as HapMap [24] and the 1000 Genomes Project [25]. It can increase the total number and density of SNPs used for association analysis and provide the opportunity to discover new potential genes.
In our study, we performed GWAS using two different genotype imputation databases and identified genetic variants related to five reproductive traits in large white pigs.
2. Materials and Methods
2.1. Ethics Statement
All ear tissue sample collection procedures were approved by the Institutional Animal Care and Use Committee of the Northwest A & F University (approval number: NWAFU-314021167).
2.2. Animals and Phenotypes
The pig population was uniformly reared at the core breeding farm of Zhumei Group Limited (Zhumadian City, China). Briefly, we collected breeding information and lineage records of large white pigs from 2011 to 2019 at this farm. There were 3733 pigs with complete pedigrees, and pedigrees could be traced back three generations. A total of 10,206 reproduction records of 2844 pigs were collected. The phenotype records included parity (including 8 levels: 1, 2, 3, 4, 5, 6, 7, or 8 or higher parity number), herd-year-season, and five reproductive traits. Five reproductive phenotypes, namely, total number born (TNB), number born alive (NBA), litter birth weight (LBW), gestation length (GL), and number of weaned (NW), were chosen for the next analysis. Table 1 presents the descriptive statistics of the five traits. Apart from GL, the other four traits had coefficients of variation above 25%.

Table 1.
Descriptive statistics for five reproductive traits.
2.3. Genotyping and Genotype Imputation
In this study, KPS Porcine Breeding 50K Chip v1 (Compass Biotechnology, Beijing, China), which contains 51,315 SNPs, was used to genotype 272 individuals of the total 2844 pigs with phenotype records. Then, quality control was performed by only keeping SNPs with MAF > 0.05, SNP call rate > 95%, individual call rate > 95%, and HWE > 1 × 10−6 using the PLINK software (v1.90) [26]. A total of 31,174 SNPs and 271 animals were retained for further GWAS. To improve the marker density, imputation was performed using two online software programs: the Pig Haplotype Reference Panel (PHARP v2) (http://alphaindex.zju.edu.cn/PHARP/index.php/, accessed on 21 October 2022) [27] and Swine Imputation Server (SWIM 1.0) (https://quantgenet.msu.edu/swim/index.html, 21 October 2022) [28]. After imputation with PHARP v2, quality control (R2 > 0.8 and MAF > 0.05) was performed, and 9,093,720 SNPs were obtained. Additionally, the SNPs were further pruned by using the “--indep-pairwise 50 5 0.9” command with a sliding window of 50 SNPs, a 5-step SNP shift, and an r2 less than 0.9. Similarly, SNPs imputed using the SWIM online software were subjected to the relevant quality control procedure. Finally, with the online imputation software PHARP and SWIM, 1,017,199 and 1,019,225 autosomal SNPs were retained.
2.4. Estimation of Genetic Parameters and Genetic Correlation
The variance and covariance components and genetic correlations of the five traits were calculated using a repeatability model in DMU v6.0 software [29].
The animal model was as follows:
In the model, y is a vector of phenotype records; b is the fixed effect of herd-year-season and parity with eight levels; X is a design matrix relating b to y; a is a vector of additive genetic effects; pe is a vector of random permanent environmental effects; and e is a vector of random residual effects. Za and Zpe are the corresponding incidence matrices.
The genetic correlation was calculated as follows:
where is the genetic correlation between trait 1 and trait 2, and represent the additive genetic values of trait 1 and trait 2 for the same individuals, and , , and refer to the genetic covariance of two traits and the genetic standard deviations of trait 1 and trait 2, respectively.
2.5. Genome-Wide Association Study (GWAS)
To perform GWAS, we used the sum of an individual’s estimated breeding value (EBV) and residual as the adjusted phenotype in this study. We used fixed and random model circulating probability unification (FarmCPU) models for GWAS in GAPIT3 [30]. This method iteratively takes advantage of the mixed linear model (MLM) as the random model and stepwise regression as the fixed model [31]. In this study, we used the Bonferroni correction method to find candidate SNPs. p < 1/N represents the genome-wide suggestive significance threshold. p < 0.05/N represents the genome-wide significance threshold. Manhattan and Q-Q plots were generated using the R CMplot package version 4.2.0 [32].
2.6. Candidate Gene Search
We used BedTools [33] to search for candidate genes in the regions 0.5 Mb downstream and upstream of the significant SNPs based on the pig reference genome (http://useast.ensembl.org/Sus_scrofa/Info/Index/, accessed on 16 December 2022, Sscrofa11.1). Additionally, to better understand the biological processes and pathways of these candidate genes, we also performed enrichment analyses. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and Gene Ontology (GO) terms were enriched via KOBAS-i [34].
3. Results
3.1. Genetic Parameters and Genetic Correlations of Reproductive Traits
The genetic parameters of the five reproductive traits are presented in Table 2. The heritability estimates of TNB, NBA, LBW, GL, and NW were 0.0442 ± 0.0011, 0.0442 ± 0.0012, 0.0476 ± 0.0025, 0.1571 ± 0.0009, and 0.0727 ± 0.0021, respectively. As can be seen, these traits are all low-heritability traits. Table 3 shows the genetic correlations of the five reproductive traits. The genetic correlations ranged from −0.235 to 0.985, with standard errors ranging from 0.001 to 0.015. Among the five reproductive traits, TNB, NBA, LBW, and NW show strong positive correlations, with correlations ranging from 0.751 to 0.985. In contrast, GL shows some negative correlations with the remaining four traits, but the correlations are not strong.
Table 2.
Estimates of variance components and genetic parameters for five reproductive traits.
Table 3.
Genetic correlations between five reproductive traits.
3.2. Identification of Significant SNPs Associated with Reproductive Traits before Imputation
In GWAS based on chip data, only three SNPs on chromosome 13 for GL reach chromosome-level significance [p < 1.60 × 10−6 (0.05/33,175)] (Figure 1A). These SNPs are located in candidate genes such as DDPA4 and DDPA2 (Table 4). Additionally, there are 5, 6, 11, and 7 SNPs that exceed the suggestive significance threshold [p < 3.01 × 10−5 (1/33,175)] for TNB, LBW, NBA, and GL, respectively (Supplementary Table S2).
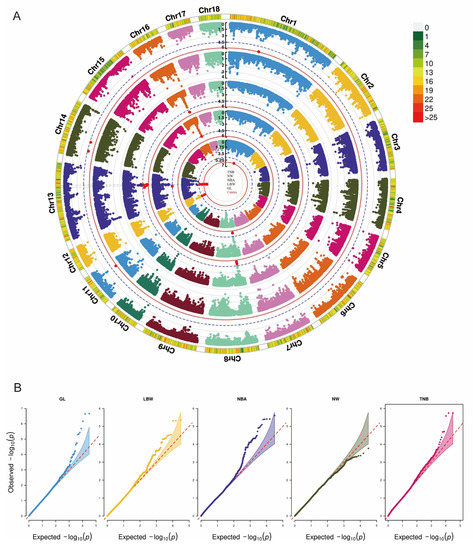
Figure 1.
Manhattan and Q-Q plots of GWAS based on chip data for five reproductive traits. (A). The red line represents the genome-wide significance level. The blue line represents the suggestive significance (3.01 × 10−5). Red spots identify SNPs with genome-wide significance (1.60 × 10−6). Traits from the inner to outer lanes are gestation length (GL), litter birth weight (LBW), number born alive (NBA), number of weaned (NW), and total number born (TNB). (B). Q-Q plots of five reproductive traits.
Table 4.
The significant SNPs in the genome for the gestation length (GL) trait using chip data in pigs.
3.3. Identification of Significant SNPs Associated with Reproductive Traits after Imputation with PHARP
Figure 2A shows the results of Manhattan plots after imputation using PHARP. The Q-Q plots are shown in Figure 2B, with genome inflation factors between 0.964 and 1.095 (Supplementary Table S1). The results show that 22 and 3 genome-wide significant SNPs [p < 4.91 × 10−8 (0.05/1,017,199)] for TNB and GL are identified, respectively (Table 5). Notably, 14 genes are identified as related to reproduction, including MRTO4, TAS1R2, PAX7, CAPZB, UBR4, KCNJ2, MITF, LDHA, LDHC, ABCC8, ARGFX, and IGSF11. According to the suggestive significance threshold [p < 9.83 × 10−7 (1/1,017,199)], 8, 3, 1, and 186 SNPs are found to be associated with TNB, NBA, LBW, and GL, respectively (Supplementary Table S3).
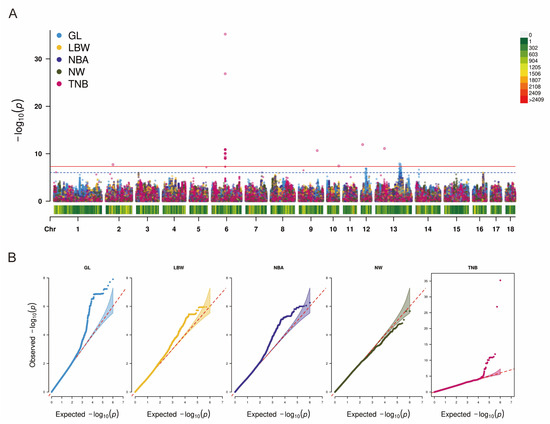
Figure 2.
Manhattan and Q-Q plots of GWAS based on data imputation using PHARP for five reproductive traits. (A) The red line represents the genome-wide significance level (4.91 × 10−8). The blue line represents the suggestive significance (9.83 × 10−7). (B) Q-Q plots of five reproductive traits. Abbreviations: GL = gestation length, LBW = litter birth weight, NBA = number born alive, NW = number of weaned, TNB = total number born.
Table 5.
The significant SNPs in the genome with the total number born (TNB) and gestation length (GL) traits using data imputed with PHARP.
3.4. Identification of Significant SNPs Associated with Reproductive Traits after Imputation with SWIM
Figure 3A shows the Manhattan plots of the genotype imputed using SWIM for GWAS. The Q-Q plots are shown in Figure 3B, with genome inflation factors between 0.889 and 1.132 (Supplementary Table S1). Overall, 271 SNPs reaching suggestive significance [p < 9.81 × 10−7 (1/1,019,225)] were found to be associated with the five reproductive traits (Supplementary Table S4). Further, 43 SNPs reach the genome-wide significance level: 8 SNPs for TNB, 3 SNPs for NBA, 8 SNPs for LBW, and 24 SNPs for GL. These significant SNPs were annotated to a number of candidate genes associated with reproduction, such as MYOCD, HMGN1, DACH1, GPC5, RPS6KA2, ARAP2, CAMK2A, and RGS18 (Table 6).
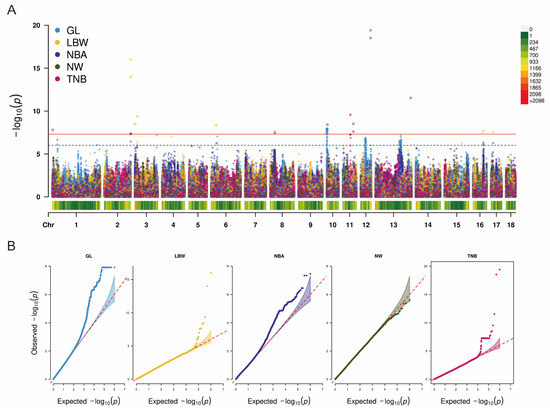
Figure 3.
Manhattan and Q-Q plots of GWAS based on data imputation with SWIM for five reproductive traits. (A) The red line represents the genome-wide significance level (4.90 × 10−8). The blue line represents the suggestive significance (9.81 × 10−7). (B) Q-Q plots of five reproductive traits. Abbreviations: GL = gestation length, LBW = litter birth weight, NBA = number born alive, NW = number of weaned, TNB = total number born.
Table 6.
The significant SNPs in the genome for the total number born (TNB) and gestation length (GL) traits using data imputed with SWIM.
3.5. Bioinformatics Annotation Analysis
In this research, candidate functional genes were found by searching 0.5 Mb upstream and downstream of the suggestive SNPs using GWAS based on chip data and two imputed databases. The genes associated with TNB are found to be linked to glycolysis/gluconeogenesis, TGF-β, the oxytocin signaling pathway, and oocyte maturation processes. For NBA and LBW, the same genes, PDGFRB, CAMK2A, and MMP2, are identified, mainly enriched in the calcium signaling pathway, GnRH signaling pathway, and embryonic organ development process. Finally, the functional genes related to GL are enriched in the mTOR signaling pathway, ovarian steroidogenesis, prolactin signaling pathway, embryo development, and regulation of G protein-coupled receptor signaling pathway (Table 7).
Table 7.
Significant KEGG pathways and GO terms associated with productive traits in pigs (p < 0.05).
4. Discussion
Reproductive traits such as TNB, NBA, LBW, GL, and NW are closely related to sow fertility and are important quantitative indicators of pig production. However, most of them have low heritability due to the complexity of the genetic structure. Therefore, it is important to clarify the genetic relationships between reproductive traits and to identify key candidate genes. In this study, a repeatability model was used to estimate the heritability of reproductive traits. The heritability estimates of the TNB, NBA, LBW, GL, and NW traits were 0.0442, 0.0442, 0.0476, 0.1571, and 0.0727, respectively. This is in agreement with the results of previous studies [35,36,37]. Additionally, we also calculated genetic correlations between individual traits and found strong positive correlations between TNB, NBA, LBW, and NW, with correlation coefficients ranging from 0.751 to 0.985, in agreement with previous reports [38,39]. This suggests that fewer traits can be selected to simplify breeding work.
Genotype imputation has been widely used in recent years with the development of sequencing technologies, price reductions, and the demand for high-density markers. This approach allows the imputation of chip data with low-density markers to WGS data, and the imputation accuracy is affected by the density of the target SNPs, the size of the reference population, the genetic distance between the target and imputation reference population, and the imputation procedure [40]. In our study, we imputed chip data using two publicly available online populating platforms. PHARP v2 provides genotype imputation using Minimac4, and the reference panel includes 4096 haplotypes, 53 million autosome SNPs, and 122 pig breeds [27]. The reference panel of SWIM 1.0 has a total of 2259 pigs, representing 44 different breeds. Based on the imputed data of the two imputation platforms mentioned above, combined with chip data, we performed GWAS for five reproductive traits.
In our study, we conducted GWAS for five reproductive traits using imputation data from two different online imputation platforms. Imputation data based on the SWIM platform detected more significant or potentially significant loci compared to the PHARP platform. This may be due to the fact that the SWIM platform has a larger number of pigs in its reference panel. In addition, an imputation strategy could improve on previous SNP-based studies without the need for additional data and expense. Furthermore, a common set of SNPs can be obtained with an imputation approach, thus making a meta-analysis possible.
Some studies have shown that the FarmCPU model can be effective in GWASs for identifying loci with low-heritability traits [35]. So, we performed GWAS by using the FarmCPU model, which divides the MLM into two parts and uses them iteratively [31]. For the TNB trait, a total of 19 suggestive candidate genes were identified based on chip data and imputed data. Among them, the RPS6KA2 gene plays a major role in the EGF signaling cascade at ovulation, which is also correlated with oocyte developmental quality [41]. As a transcription factor, SMAD4 plays an important role in the porcine reproductive system. It has been shown that miR-143 [42], miR-26b [43], and miR-10b [44] can inhibit apoptosis and promote E2 release via SMAD4 in porcine granulosa cells. For both the NBA and LBW traits, GWAS based on imputed data identified the CAMK2A, NDST1, and RPS14 genes. In a meta-analysis of reproductive traits in heifers, the CAMK2A gene was identified as being involved in calcium signaling mechanisms and acting on pituitary gonadotropin secretion [45]. This is consistent with our findings. In addition, NDST1 has been shown to be critical for many organogenesis processes, and the targeted disruption of the NDST1 gene impaired heart development in mice [46]. NSDT1 f/f/2 null/3 null mice with defective decidualization resulted in female infertility [47]. It has been reported that RSP14 is a key gene in early embryonic development [48]. Embryonic stem cells heterozygous for the RSP14 gene showed defects in embryoid body differentiation [49]. For the GL trait, both GWASs based on chip data and imputed data identified genome-wide significant SNPs. Based on KEGG and GO analyses, we annotated a total of 13 candidate genes, mainly related to the ovarian steroidogenesis pathway and embryo development process. Among these, ADCY5 was identified as being associated with seasonal estrus in Sunite sheep [50], egg production in white Muscovy ducks [51], and fertility in cows [52], while in human GWAS, ADCY5 was found to be associated with gestational duration [53]. Interestingly, it has been reported that ADCY5 is associated with fetal growth and birth weight [54]. However, the ADCY5 gene has not been studied in pig reproduction, and we speculate that this gene may be a key gene in the influence of reproductive performance in pigs. Unfortunately, no potential SNPs were identified for the NW trait, probably due to the small size of the population and the high number of missing phenotypic data points. Overall, our results identify a number of new key candidate genes and loci associated with reproductive traits in large white pigs, but further studies are needed to confirm the functions of these genes.
5. Conclusions
In this study, the genetic parameters of TNB, NBA, LBW, GL, and NW in Yorkshire pigs were estimated using a repeatability model. These traits are low-heritability traits. There were strong positive correlations between TNB, NBA, LBW, and NW, excluding the GL trait, which was weakly negatively correlated with them. GWASs based on chip data and imputed data were performed for five reproductive traits in Yorkshire pigs. Finally, combining the results of GWAS and bioinformatics annotation analysis, SMAD4, RPS6KA2, CAMK2A, NDST1, and ADCY5 were identified as novel genes, and some of them have not been studied in livestock, so they may be key candidate genes affecting reproductive traits in pigs. The results of this study highlight some new major genes regulating reproductive traits in pigs and can benefit genome selection for pig genetic breeding.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/genes14040861/s1, Table S1. The genomic inflation factor (λ) for each GWAS using chip and imputation data of pigs. Table S2. Genes within 0.5 Mb of suggestive significant SNPs identified by GWAS based on chip data for reproductive traits. Table S3. Genes within 0.5 Mb of suggestive significant SNPs identified by GWAS based on the PHARP imputation data for reproductive traits. Table S4. Genes within 0.5 Mb of suggestive significant SNPs identified by GWAS based on SWIM imputation data for reproductive traits.
Author Contributions
G.Y. and X.D. conceived and supervised the study. Y.J., M.C., and J.W. helped complete the imputation of the chip data. T.Y. provided technical guidance. J.X. collected the samples and recorded the phenotypes. Y.W. extracted the DNA for genotyping. J.S. analyzed the data and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The authors are grateful to all the funding agencies. This work was supported by the National Key R&D Program of China (2021YFF1000602, 2021YFD1301200) and China Agriculture Research System (CARS-35-PIG).
Institutional Review Board Statement
The whole collection procedure of ear tissue samples was approved by the Institutional Animal Care and Use Committee of the Northwest A & F University (approval number: NWAFU-314021167).
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Sell-Kubiak, E.; Knol, E.F.; Lopes, M. Evaluation of the phenotypic and genomic background of variability based on litter size of Large White pigs. Genet. Sel. Evol. 2022, 54, 1. [Google Scholar] [CrossRef]
- Akanno, E.C.; Schenkel, F.S.; Sargolzaei, M.; Friendship, R.M.; Robinson, J.A. Opportunities for genome-wide selection for pig breeding in developing countries. J. Anim. Sci. 2013, 91, 4617–4627. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bosse, Y.; Pare, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Liu, H.T.; Song, H.L.; Jiang, Y.F.; Jiang, Y.; Zhang, F.X.; Liu, Y.B.; Shi, Y.; Ding, X.D.; Wang, C.D. A Single-Step Genome Wide Association Study on Body Size Traits Using Imputation-Based Whole-Genome Sequence Data in Yorkshire Pigs. Front. Genet. 2021, 12, 629049. [Google Scholar] [CrossRef]
- Li, J.; Peng, S.; Zhong, L.P.; Zhou, L.S.; Yan, G.R.; Xiao, S.J.; Ma, J.W.; Huang, L.S. Identification and validation of a regulatory mutation upstreAm. of the BMP2 gene associated with carcass length in pigs. Genet. Sel. Evol. 2021, 53, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.F.; Zhang, J.J.; Gong, H.F.; Cui, L.L.; Zhang, W.C.; Ma, J.W.; Chen, C.Y.; Ai, H.S.; Xiao, S.J.; Huang, L.S.; et al. Genetic correlation of fatty acid composition with growth, carcass, fat deposition and Meat. quality traits based on GWAS data in six pig populations. Meat. Sci. 2019, 150, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.X.; Zhang, J.J.; Xiong, X.W.; Chen, C.Y.; Xing, Y.Y.; Duan, Y.Y.; Xiao, S.J.; Yang, B.; Ma, J.W. An Integrative Analysis of Transcriptome and GWAS Data to Identify Potential Candidate Genes Influencing Meat. Quality Traits in Pigs. Front. Genet. 2021, 12, 748070. [Google Scholar] [CrossRef]
- Delpuech, E.; Aliakbari, A.; Labrune, Y.; Feve, K.; Billon, Y.; Gilbert, H.; Riquet, J. Identification of genomic regions affecting production traits in pigs divergently selected for feed efficiency. Genet. Sel. Evol. 2021, 53, 1–5. [Google Scholar] [CrossRef]
- Silva, E.F.; Lopes, M.S.; Lopes, P.S.; Gasparino, E. A genome-wide association study for feed efficiency-related traits in a crossbred pig population. Animal 2019, 13, 2447–2456. [Google Scholar] [CrossRef]
- Marques, D.B.D.; Bastiaansen, J.W.M.; Broekhuijse, M.L.W.J.; Lopes, M.S.; Knol, E.F.; Harlizius, B.; Guimaraes, S.E.F.; Silva, F.F.; Lopes, P.S. Weighted single-step GWAS and gene network analysis reveal new candidate genes for semen traits in pigs. Genet. Sel. Evol. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Chen, Y.L.; Liu, X.H.; Zhao, Y.X.; Zhu, L.; Liu, A.; Jiang, W.; Peng, X.; Zhang, C.L.; Tang, Z.S.; et al. Weighted single-step GWAS identified candidate genes associated with semen traits in a Duroc boar population. BMC Genomics 2019, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Y.; Fu, Y.H.; Hu, Y.; Yin, L.L.; Tang, Z.S.; Yin, D.; Zhu, M.J.; Yu, M.; Li, X.Y.; Zhou, Y.; et al. Whole genome variants across 57 pig breeds enable comprehensive identification of genetic signatuRes. that underlie breed features. J. Anim. Sci. Biotechno. 2020, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.P.; Deng, Z.; Huang, M.; Hou, Y.; Zhang, H.; Chen, H.; Ren, J. Whole-Genome Resequencing Identifies KIT New Alleles That Affect Coat Color Phenotypes in Pigs. Front. Genet. 2019, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- Sevillano, C.A.; Lopes, M.S.; Harlizius, B.; Hanenberg, E.H.A.T.; Knol, E.F.; Bastiaansen, J.W.M. Genome-wide association study using deregressed breeding values for cryptorchidism and scrotal/inguinal hernia in two pig lines. Genet. Sel. Evol. 2015, 47, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Flossmann, G.; Wurmser, C.; Pausch, H.; Tenghe, A.; Dodenhoff, J.; Dahinten, G.; Gotz, K.U.; Russ, I.; Fries, R. A nonsense mutation of bone morphogenetic protein-15 (BMP15) causes both infertility and increased litter size in pigs. BMC Genomics 2021, 22, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.K.; Huang, T.; Zhang, M.P.; Chen, J.Q.; Zhang, Z.; Li, J.Q.; Du, H.P.; Ling, Z.Q.; Wu, Z.Z.; Yang, B.; et al. Four genetic loci affecting swine lung lesions identified by whole-genome sequencing-based association studies. Sci. China Life Sci. 2021, 64, 1571–1574. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Huang, T.; Deng, W.; Yan, G.; Qiu, H.; Huang, Y.; Ke, S.; Hou, Y.; Zhang, Y.; Zhang, Z.; et al. Genome-wide association studies identify susceptibility loci affecting respiratory disease in Chinese Erhualian pigs under natural conditions. Anim. Genet. 2017, 48, 30–37. [Google Scholar] [CrossRef]
- Reverter, A.; Ballester, M.; Alexandre, P.A.; Marmol-Sanchez, E.; Dalmau, A.; Quintanilla, R.; Ramayo-Caldas, Y. A gene co-association network regulating gut microbial communities in a Duroc pig population. Microbiome 2021, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef]
- Li, M.Z.; Chen, L.; Tian, S.L.; Lin, Y.; Tang, Q.Z.; Zhou, X.M.; Li, D.Y.; Yeung, C.K.L.; Che, T.D.; Jin, L.; et al. Comprehensive variation discovery and recovery of missing sequence in the pig genome using multiple de novo assemblies. Genome Res. 2017, 27, 865–874. [Google Scholar] [CrossRef]
- Ai, H.S.; Fang, X.D.; Yang, B.; Huang, Z.Y.; Chen, H.; Mao, L.K.; Zhang, F.; Zhang, L.; Cui, L.L.; He, W.M.; et al. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nat. Genet. 2015, 47, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Z.; Tian, S.L.; Jin, L.; Zhou, G.Y.; Li, Y.; Zhang, Y.; Wang, T.; Yeung, C.K.L.; Chen, L.; Ma, J.D.; et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 2013, 45, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Marchini, J.; Howie, B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010, 11, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Ballinger, D.G.; Cox, D.R.; Hinds, D.A.; Stuve, L.L.; Gibbs, R.A.; Belmont, J.W.; Boudreau, A.; Hardenbol, P.; Leal, S.M.; et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007, 449, 851–853. [Google Scholar] [CrossRef]
- Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; Gabriel, S.B.; et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Z.; Chen, Z.; Sun, J.; Cao, C.; Wu, F.; Xu, Z.; Zhao, W.; Sun, H.; Guo, L.; et al. PHARP: A pig haplotype reference panel for genotype imputation. Sci. Rep. 2022, 12, 12645. [Google Scholar] [CrossRef]
- Ding, R.; Savegnago, R.; Liu, J.; Long, N.; Tan, C.; Cai, G.; Zhuang, Z.; Wu, J.; Yang, M.; Qiu, Y.; et al. Nucleotide resolution genetic mapping in pigs by publicly accessible whole genome imputation. bioRxiv 2022, 2022, 2022-05. [Google Scholar] [CrossRef]
- Madsen, P., Sørensen, P., Su, G., Damgaard, L., Thomsen, H., Labouriau, R., Eds.; DMU-a package for analyzing multivariate mixed models. In Proceedings of the 8th World Congress on Genetics Applied to Livestock Production, Belo Horizonte, Brazil, 13–16 August 2006. [Google Scholar]
- Wang, J.; Zhang, Z. GAPIT Version 3: Boosting Power and Accuracy for Genomic Association and Prediction. Genomics Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M.J.B. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.X.; Gao, G.X.; Zhou, Y.; Guo, C.X.; Li, B.; El-Ashram, S.; Li, Z.L. Genome-wide association studies uncover genes associated with litter traits in the pig. Animal 2022, 16, 100672. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, Z.; Ye, S.; He, Y.; Huang, S.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J. Genome-Wide Association Study for Reproductive Traits in a Duroc Pig Population. Animals 2019, 9, 732. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Li, H.; Wang, L. Genetic parameters estimation and genome molecular marker identification for gestation length in pigs. Front. Genet. 2022, 13, 1046423. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, Y.; Huang, X.; Wu, S.; Bao, W. A genome-wide association study of important reproduction traits in large white pigs. Gene 2022, 838, 146702. [Google Scholar] [CrossRef]
- Zhang, T.; WANG, L.-g.; SHI, H.-b.; Hua, Y.; ZHANG, L.-c.; Xin, L.; Lei, P.; Liang, J.; ZHANG, Y.-b.; ZHAO, K.-b.J.J.o.I.A. Hritabilities and genetic and phenotypic correlations of litter uniformity and litter size in Large White sows. Animal 2016, 15, 848–854. [Google Scholar] [CrossRef]
- Deng, T.; Zhang, P.; Garrick, D.; Gao, H.; Wang, L.; Zhao, F. Comparison of Genotype Imputation for SNP Array and Low-Coverage Whole-Genome Sequencing Data. Front. Genet. 2021, 12, 704118. [Google Scholar] [CrossRef]
- Adriaenssens, T.; Wathlet, S.; Segers, I.; Verheyen, G.; De Vos, A.; Van der Elst, J.; Coucke, W.; Devroey, P.; Smitz, J. Cumulus cell gene expression is associated with oocyte developmental quality and influenced by patient and treatment characteristics. Hum. Reprod 2010, 25, 1259–1270. [Google Scholar] [CrossRef]
- Du, X.; Zhang, L.; Li, X.; Pan, Z.; Liu, H.; Li, Q. TGF-beta signaling controls FSHR signaling-reduced ovarian granulosa cell apoptosis through the SMAD4/miR-143 axis. Cell Death Dis. 2016, 7, e2476. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Du, X.; Zhou, J.; Pan, Z.; Liu, H.; Li, Q. MicroRNA-26b functions as a proapoptotic factor in porcine follicular Granulosa cells by targeting Sma-and Mad-related protein 4. Biol. Reprod 2014, 91, 146. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Du, X.; Pan, Z.; Zhang, L.; Li, Q. The transcription factor SMAD4 and miR-10b contribute to E2 release and cell apoptosis in ovarian granulosa cells by targeting CYP19A1. Mol. Cell Endocrinol. 2018, 476, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M.S.; Porto-Neto, L.R.; Gondro, C.; Shittu, O.B.; Wockner, K.; Tan, A.W.L.; Smith, H.R.; Gouveia, G.C.; Kour, J.; Fortes, M.R.S. Meta-Analysis of Heifer Traits Identified Reproductive Pathways in Bos indicus Cattle. Genes 2021, 12, 768. [Google Scholar] [CrossRef]
- Pan, Y.; Carbe, C.; Kupich, S.; Pickhinke, U.; Ohlig, S.; Frye, M.; Seelige, R.; Pallerla, S.R.; Moon, A.M.; Lawrence, R.; et al. Heparan sulfate expression in the neural crest is essential for mouse cardiogenesis. Matrix Biol. 2014, 35, 253–265. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, A.; Feng, L.; Wang, Y.; Zhang, H.; Zhang, I.; Bany, B.M.; Ma, L. Heparan Sulfate Proteoglycan Sulfation Regulates Uterine Differentiation and Signaling During Embryo Implantation. Endocrinology 2018, 159, 2459–2472. [Google Scholar] [CrossRef]
- Liu, H.B.; Muhammad, T.; Guo, Y.; Li, M.J.; Sha, Q.Q.; Zhang, C.X.; Liu, H.; Zhao, S.G.; Zhao, H.; Zhang, H.; et al. RNA-Binding Protein IGF2BP2/IMP2 is a Critical Maternal Activator in Early Zygotic Genome Activation. Adv Sci. 2019, 6, 1900295. [Google Scholar] [CrossRef]
- Fortier, S.; MacRae, T.; Bilodeau, M.; Sargeant, T.; Sauvageau, G. Haploinsufficiency screen highlights two distinct groups of ribosomal protein genes essential for embryonic stem cell fate. Proc Natl Acad Sci. USA 2015, 112, 2127–2132. [Google Scholar] [CrossRef]
- Du, X.; He, X.; Liu, Q.; Di, R.; Liu, Q.; Chu, M. Comparative Transcriptomics Reveals the Key lncRNA and mRNA of Sunite Sheep Adrenal Gland Affecting Seasonal Reproduction. Front. Vet. Sci. 2022, 9, 816241. [Google Scholar] [CrossRef]
- Bello, S.F.; Xu, H.; Guo, L.; Li, K.; Zheng, M.; Xu, Y.; Zhang, S.; Bekele, E.J.; Bahareldin, A.A.; Zhu, W.; et al. Hypothalamic and ovarian transcriptome profiling reveals potential candidate genes in low and high egg production of white Muscovy ducks (Cairina moschata). Poult. Sci. 2021, 100, 101310. [Google Scholar] [CrossRef]
- Li, J.; Shen, C.; Zhang, K.; Niu, Z.; Liu, Z.; Zhang, S.; Wang, Y.; Lan, X. Polymorphic variants of bovine ADCY5 gene identified in GWAS analysis were significantly associated with ovarian morphological related traits. Gene 2021, 766, 145158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Feenstra, B.; Bacelis, J.; Liu, X.; Muglia, L.M.; Juodakis, J.; Miller, D.E.; Litterman, N.; Jiang, P.P.; Russell, L.; et al. Genetic Associations with Gestational Duration and Spontaneous Preterm Birth. N. Engl. J. Med. 2017, 377, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Freathy, R.M.; Mook-Kanamori, D.O.; Sovio, U.; Prokopenko, I.; Timpson, N.J.; Berry, D.J.; Warrington, N.M.; Widen, E.; Hottenga, J.J.; Kaakinen, M.; et al. Variants in ADCY5 and near CCNL1 are associated with fetal growth and birth weight. Nat. Genet. 2010, 42, 430–435. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).