
Description of a Cohort with a New Truncating MYBPC3 Variant for Hypertrophic Cardiomyopathy in Northern Spain
 , ,
, , 
Abstract
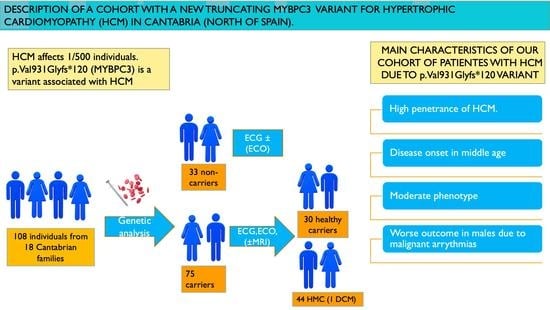
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Graphic Representation of the Data Included in Table 1



Appendix B. Table of Additional Genetic Variants Mentioned

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Variant | RS Number | Clinical Significance | Frequency in General Population | Cited by |
|---|---|---|---|---|---|
| RBM20 | NP_001127835.2:p.Asp888Asn | rs201370621 | Unknown significance (VUS) | Rare variant (present in <1% of controls) | Refaat et al. 2012 [45] |
| MYL2 | NM_000432.4:c.354-3C>G | rs758158083 | Unknown significance (VUS) | Not present in controls. | Restrepo-Córdoba et al. 2017 [46] |
| MYH6 | NP_002462.2:p.Leu120Phe | rs769074801 | Unknown significance (VUS) | Not present in controls. | |
| TNNT2 | NP_001001430.1:p.Arg286His | rs141121678 | Pathogenic, associated with the development of hypertrophic cardiomyopathy (HCM). | 0.007% | Ripoll-Vera et al. 2016 [23] |
References
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell 1999, 62, 99–1006. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Navarro-López, F. Hypertrophic cardiomyopathy. Genetic basis and clinical implications. Rev. Esp. Cardiol. 2004, 57 (Suppl. S1), 22–32. [Google Scholar] [CrossRef] [PubMed]
- Elliot, P.; McKenna, W.J. Hypertrophic cardiomyopathy. Lancet 2004, 363, 1881–1891. [Google Scholar] [CrossRef]
- Semsarian, C.; CSANZ Cardiovascular Genetics Working Group. Guidelines por the diagnosis and management of hypertrophic cardiomyopathy. Heart Lung Circ. 2007, 16, 16–18. [Google Scholar] [CrossRef]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. EUROGENE Heart Failure Project. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M. Genetic evaluation of cardiomyopathy—A Heart Failure Society of America Practice Guideline. J. Card. Fail. 2018, 24, 281–302. [Google Scholar] [CrossRef]
- Navarro-López, F. Hypertrophic cardiomyopathy: Never-ending complexity. Rev. Esp. Cardiol. 2006, 59, 994–996. [Google Scholar] [CrossRef]
- Carrier, L.; Bonne, G.; Bährend, E.; Yu, B.; Richard, P.; Niel, F.; Hainque, B.; Cruaud, C.; Gary, F.; Labeit, S.; et al. Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ. Res. 1997, 80, 427–434. [Google Scholar] [CrossRef]
- Méndez, I.; Fernández, A.I.; Espinosa, M.A.; Cuenca, S.; Lorca, R.; Rodríguez, J.F.; Tamargo, M.; García-Montero, M.; Gómez, C.; Vilches, S.; et al. Founder mutation in myosin-binding protein C with an early onset and a high penetrance in males. Open. Heart. 2021, 8, e001789. [Google Scholar] [CrossRef]
- Wessels, M.W.; Herkert, J.C.; Frohn-Mulder, I.M.; Dalinghaus, M.; van den Wijngaard, A.; de Krijger, R.R.; Michels, M.; de Coo, I.F.M.; Hoedemaekers, Y.M.; Dooijes, D. Compound heterozygous or homozygous truncating MYBPC3 mutations cause lethal cardiomyopathy with features of noncompaction and septal defects. Eur. J. Hum. Genet. 2015, 23, 922–928. [Google Scholar] [CrossRef]
- Oliva-Sandoval, M.J.; Ruiz-Espejo, F.; Monserrat, L.; Hermida-Prieto, M.; Sabater, M.; García-Molina, E.; Ortiz, M.; Rodríguez-García, M.I.; Núñez, L.; Gimeno, J.R.; et al. Insights into genotype-phenotype correlation in hypertrophic cardiomyopathy. Findings from 18 Spanish families with a single mutation in MYBPC3. Heart 2010, 96, 1980–1984. [Google Scholar] [CrossRef]
- Charron, P.; Dubourg, O.; Desnos, M.; Bennaceur, M.; Carrier, L.; Camproux, A.C.; Isnard, R.; Hagege, A.; Langlard, J.M.; Bonne, G.; et al. Clinical features and prognostic implications of familial hypertrophic cardiomyopathy related to the cardiac myosin-binding protein C gene. Circulation 1998, 97, 2230–2236. [Google Scholar] [CrossRef]
- Konno, T.; Shimizu, M.; Ino, H.; Matsuyama, T.; Yamaguchi, M.; Terai, H.; Hayashi, K.; Mabuchi, T.; Kiyama, M.; Sakata, K.; et al. A novel missense mutation in the myosin binding protein-C gene is responsible for hypertrophic cardiomyopathy with left ventricular dysfunction and dilation in elderly patients. J. Am. Coll. Cardiol. 2003, 41, 781–786. [Google Scholar] [CrossRef]
- Van Driest, S.L.; Vasile, V.C.; Ommen, S.R.; Will, M.L.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 1903–1910. [Google Scholar] [CrossRef]
- Morita, H.; Rehm, H.L.; Menesses, A.; McDonough, B.; Roberts, A.E.; Kucherlapati, R.; Towbin, J.A.; Seidman, J.G.; Seidman, C.E. Shared genetic causes of cardiac hypertrophy in children and adults. N. Engl. J. Med. 2008, 358, 1899–1908. [Google Scholar] [CrossRef]
- Reguero, J.R.; Gómez, J.; Martín, M.; Flórez, J.P.; Morís, C.; Iglesias, S.; Alonso, B.; Álvarez, V.; Coto, E. The G263X MYBPC3 mutation is a common and low-penetrant mutation for hypertrophic cardiomyopathy in the region of Asturias (Northern Spain). Int. J. Cardiol. 2013, 168, 4555–4556. [Google Scholar] [CrossRef]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart., J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef]
- Maron, B.J. Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Circulation 2010, 121, 445–456. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Rupp, S.; Felimban, M.; Schänzer, A.; Schranz, D.; Marschall, C.; Zenker, M.; Logeswaran, T.; Neuhäuser, C.; Thul, J.; Jux, C.; et al. Genetic basis of hypertrophic cardiomyopathy in children. Clin. Res. Cardiol. 2019, 108, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Li, Z.; Huang, W.; Chen, P.; Sun, Y.; Wang, H.; Wu, D.; Chen, Y.; Li, C.; Xiao, L.; et al. RBM20 is a candidate gene for Hypertrophic Cardiomyopathy. Can. J. Cardiol. 2021, 37, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Ripoll-Vera, T.; Gámez, J.M.; Govea, N.; Gómez, Y.; Núñez, J.; Socías, L.; Escandell, A.; Rosell, J. Clinical and prognostic profiles of cardiomyopathies caused by mutations in the Troponin T gene. Rev. Esp. Cardiol. 2016, 69, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, T.; Robinet, S.; Dulgheru, R.; Bernard, A.; Ilardi, F.; Contu, L.; Addetia, K.; Caballero, L.; Kacharava, G.; Athanassopoulos, G.D.; et al. Echocardiographic reference ranges for normal left atrial function parameters: Results from the EACVI NORRE study. Eur. Heart J. Cardiovasc. Imaging 2018, 19, 630–638. [Google Scholar] [CrossRef]
- Gómez, J.; Reguero, J.R.; Morís, C.; Martín, M.; Álvarez, V.; Alonso, B.; Iglesias, S.; Coto, E. Mutation analysis of the main hypertrophic cardiomyopathy genes using multiplex amplification and semiconductor next-generation sequencing. Circ. J. 2014, 78, 2963–2971. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Zhou, X.; Song, L. Progress in the molecular genetics of hypertrophic cardiomyopathy: A mini-review. Gerontology 2013, 59, 199–205. [Google Scholar] [CrossRef]
- Schlossarek, S.; Mearini, G.; Carrier, L. Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: Mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2011, 50, 613–620. [Google Scholar] [CrossRef]
- Sabater-Molina, M.; Saura, D.; García-Molina Sáez, E.; González-Carrillo, J.; Polo, L.; Pérez-Sánchez, I.; Olmo, M.C.; Oliva-Sandoval, M.J.; Barriales-Villa, R.; Carbonell, P.; et al. A novel founder mutation in MYBPC3: Phenotypic comparison with the most prevalent MYBPC3 mutation in Spain. Rev. Esp. Cardiol. 2017, 70, 105–114. [Google Scholar] [CrossRef]
- Kubo, T.; Kitaoka, H.; Okawa, M.; Matsumura, Y.; Hitomi, N.; Yamasaki, N.; Furuno, T.; Takata, J.; Nishinaga, M.; Kimura, A.; et al. Lifelong left ventricular remodeling of hypertrophic cardiomyopathy caused by a founder frameshift deletion mutation in the cardiac Myosin-binding protein C gene among Japanese. J. Am. Coll. Cardiol. 2005, 46, 1737–1743. [Google Scholar] [CrossRef]
- Spirito, P.; Bellone, P.; Harris, K.M.; Bernabo, P.; Bruzzi, P.; Maron, B.J. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N. Engl. J. Med. 2000, 342, 1778–1785. [Google Scholar] [CrossRef]
- Siontis, K.C.; Geske, J.B.; Ong, K.; Nishimura, R.A.; Ommen, S.R.; Gersh, B.J. Atrial fibrillation in hypertrophic cardiomyopathy: Prevalence, clinical correlations, and mortality in a large high-risk population. J. Am. Heart Assoc. 2014, 3, e001002. [Google Scholar] [CrossRef]
- Terauchi, Y.; Kubo, T.; Baba, Y.; Hirota, T.; Tanioka, K.; Yamasaki, N.; Furuno, T.; Kitaoka, H. Gender differences in the clinical features of hypertrophic cardiomyopathy caused by mutations in the cardiac myosin-binding protein C gene. J. Cardiol. 2015, 65, 423–428. [Google Scholar] [CrossRef]
- Lorca, R.; Gómez, J.; Martín, M.; Cabanillas, R.; Calvo, J.; León, V.; Pascual, I.; Morís, C.; Coto, E.; Reguero, J.J.R. Insights into hypertrophic cardiomyopathy evaluation through follow-up of a founder pathogenic variant. Rev. Esp. Cardiol. 2019, 72, 138–144. [Google Scholar] [CrossRef]
- Frisso, G.; Limongelli, G.; Pacileo, G.; Del Giudice, A.; Forgione, L.; Calabrò, P.; Iacomino, M.; Detta, N.; Di Fonzo, L.M.; Maddaloni, V.; et al. A child cohort study from southern Italy enlarges the genetic spectrum of hypertrophic cardiomyopathy. Clin. Genet. 2009, 76, 91–101. [Google Scholar] [CrossRef]
- Pelliccia, A.; Maron, M.S.; Maron, B.J. Assessment of left ventricular hypertrophy in a trained athlete: Differential diagnosis of physiologic athlete’s heart from pathologic hypertrophy. Prog. Cardiovasc. Dis. 2012, 54, 387–396. [Google Scholar] [CrossRef]
- Snir, A.W.; Connelly, K.A.; Goodman, J.M.; Dorian, D.; Dorian, P. Exercise in hypertrophic cardiomyopathy: Restrict or rethink. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, 2101–2111. [Google Scholar] [CrossRef]
- Maron, B.J.; Udelson, J.E.; Bonow, R.O.; Nishimura, R.A.; Ackerman, M.J.; Estes, N.A.M., 3rd; Cooper, L.T., Jr.; Link, M.S.; Maron, M.S. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task force 3: Hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy and other cardiomyopathies, and myocarditis: A scientific statement from the American Heart Association and American College of Cardiology. J. Am. Coll. Cardiol. 2015, 66, 2362–2371. [Google Scholar] [CrossRef]
- Gimeno, J.R.; Tomé-Esteban, M.; Lofiego, C.; Hurtado, J.; Pantazis, A.; Mist, B.; Lambiase, P.; McKenna, W.J.; Elliott, P.M. Exercise-induced ventricular arrhythmias and risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Eur. Heart J. 2009, 30, 2599–2605. [Google Scholar] [CrossRef]
- Wang, H.; Lin, Y.; Zhang, R.; Chen, Y.; Ji, W.; Li, S.; Wang, L.; Tan, R.; Yuan, J. Programmed exercise attenuates familial hypertrophic cardiomyopathy in transgenic E22K mice via inhibition of PKC-α/NFAT pathway. Front. Cardiovasc. Med. 2022, 9, 808163. [Google Scholar] [CrossRef]
- McMullen, J.R.; Amirahmadi, F.; Woodcock, E.A.; Schinke-Braun, M.; Bouwman, R.D.; Hewitt, K.A.; Mollica, J.P.; Zhang, L.; Zhang, Y.; Shioi, T.; et al. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2007, 104, 612–617. [Google Scholar] [CrossRef]
- Dejgaard, L.A.; Haland, T.F.; Lie, O.H.; Ribe, M.; Bjune, T.; Leren, I.S.; Berge, K.E.; Edvardsen, T.; Haugaa, K.H. Vigorous exercise in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2018, 250, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Maca-Meyer, N.; Sánchez-Velasco, P.; Flores, C.; Larruga, J.M.; González, A.M.; Oterino, A.; Leyva-Cobián, F. Y chromosome and mitochondrial DNA characterization of Pasiegos, a human isolate from Cantabria (Spain). Ann. Hum. Genet. 2003, 67, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.; Zarrabeitia, M.T.; Valverde, L.; Odriozola, A.; Alfonso-Sánchez, M.A.; de Pancorbo, M.M. Variability of the entire mitochondrial DNA control region in a human isolate from the Pas Valley (northern Spain). J. Forensic. Sci. 2010, 55, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J. On genetic and phenotypic variability of hypertrophic cardiomyopathy: Nature versus nurture. J. Am. Coll. Cardiol. 2001, 38, 331–334. [Google Scholar] [CrossRef]
- Refaat, M.M.; Lubitz, S.A.; Makino, S.; Islam, Z.; Frangiskakis, J.M.; Mehdi, H.; Gutmann, R.; Zhang, M.L.; Bloom, H.L.; MacRae, C.A.; et al. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm. 2012, 9, 390–396. [Google Scholar] [CrossRef]
- Restrepo-Cordoba, M.A.; Campuzano, O.; Ripoll-Vera, T.; Cobo-Marcos, M.; Mademont-Soler, I.; Gámez, J.M.; Fernando Dominguez, J.M.; Gonzalez-Lopez, E.; Padron-Barthe, L.; Lara-Pezzi, E.; et al. Usefulness of Genetic Testing in Hypertrophic Cardiomyopathy: An Analysis Using Real-World Data. J. Cardiovasc. Transl. Res. 2017, 10, 35–46. [Google Scholar] [CrossRef]





| Carrying Patients with HCM | ||||
|---|---|---|---|---|
| All | Men | Women | p | |
| N | 44 | 28 | 16 | |
| Current age + | 55 ± 17 | 56 ± 16 | 55 ± 20 | 0.846 |
| Diagnosis age | 41 ± 17 | 39 ± 15 | 46 ± 19 | 0.206 |
| Reason for diagnosis: | 0.382 | |||
| Symptoms | 20 (45.5%) | 14 (50%) | 6 (37.5%) | |
| Family history of disease | 19 (43.2%) | 10 (35.7%) | 9 (56.3%) | |
| Irregular ECG | 5 (11.4%) | 4 (14.3%) | 1 (6.3%) | |
| Thickness of left ventricle wall (IVS) | 20.5 ± 5.8 (Range 13.0–43.0) | 21.4 ± 6.3 (Range 13.0–43.0) | 18.9 ± 4.6 (Range 13.0–26.0) | 0.208 |
| Thickness of posterior wall (LVPW) | 12.2 ± 4.5 (Range 6.6–30.0) | 12.3 ± 4.8 (Range 7.6–30.0) | 11.8 ± 3.9 (Range 6.6–20.0) | 0.759 |
| Hypertrophy type: | 0.346 | |||
| Apical | 1 (2.3%) | 0 (0%) | 1 (6.3%) | |
| Asymmetric septal | 31 (70.5%) | 21 (75%) | 10 (62.5%) | |
| Concentric | 12 (27.3%) | 7 (25%) | 5 (31.3%) | |
| Irregular ECG | 37/7 (84.1%) | 24/4 (85.7%) | 13/3 (81.2%) | 0.3524 |
| Symptoms: | 24/20 (54.5%) | 16/12 (57.1%) | 8/8 (50%) | 0.647 |
| Breathlessness | 19(43.2%) | 14 (50%) | 5 (31.3%) | 0.227 |
| Chest pain | 6 (13.6%) | 3 (10.7%) | 3 (18.8%) | 0.455 |
| Palpitations | 6 (13.6%) | 3 (10.7%) | 3 (18.8%) | 0.455 |
| Syncope | 5 (11.4%) | 4 (14.3%) | 1 (6.3%) | 0.419 |
| NYHA class | 0.897 | |||
| I + II | 33 (75%) | 20 (71.4%) | 13 (812%) | |
| III + IV | 11 (25%) | 8 (28.6%) | 3 (18.7%) | |
| Obstruction | 11/33 (25%) | 7/21 (25%) | 4/12 (25%) | 1 |
| Systolic dysfunction | 7/37 (15.9%) | 6/22 (21.4%) | 1/15 (6.3%) | 0.185 |
| Diastolic dysfunction | 33/11 (75%) | 23/5 (82.1%) | 10/6 (62.5%) | 0.148 |
| Left atrial (LA) diameter | ||||
| Normal < 45 mm | 22 (50%) | 15 (53.6%) | 7 (43.8%) | |
| Dilated ≥ 45 mm | 22 (50%) | 13 (46.4%) | 9 (56.2%) | 0.531 |
| Atrial fibrillation (AF) | 15/29 (34.1%) | 8/20 (28.6%) | 7/9 (43.8%) | 0.307 |
| Arrhythmia | 27/17 (61.4%) | 17/11 (60.7%) | 10/6 (62.5%) | 0.907 |
| Non-sustained tachycardia in Holter monitoring (NSVT) | 12/32 (27.3%) | 11/17 (39.3%) | 1/15 (8.3%) | 0.018 |
| Fibrosis in magnetic resonance imaging (MRI) | 16/12 (57.1%) | 12/5 (70.6%) | 4/7 (36.4 %) | 0.074 |
| Semi-professional sport | 11/33 (25%) | 11/17 (39.3%) | 0/16 (0%) | 0.004 |
| Implantable cardioverter defibrillator (ICD) | 18/26 (40.9%) | 15/13 (53.6%) | 3/13 (18.8%) | 0.024 |
| Primary prevention | 14 (77.8%) | 11 (73.3%) | 3 (100%) | |
| Secondary prevention | 4 (22.2%) | 4 (26.7%) | 0 (0%) | 0.310 |
| Other procedures: | ||||
| Heart transplant | 2 (4.5%) | 2 (7.1%) | 0 (0%) | |
| Myomectomy | 3 (6.8%) | 1 (3.6%) | 2 (12.5%) | |
| Alcohol septal ablation (ASA) | 1 (2.3%) | 0 (0%) | 1 (6.2%) | |
| Total events: | 9 (20.4%) | 6 (21.4%) | 4 (25%) | 0.6056 |
| Acute myocardial infarction (AMI) | 1 (2.3%) | 1 (3.6%) | 0 (0%) | |
| Stroke | 2 (4.5%) | 1 (3.6%) | 1 (6.2%) | |
| Cardiac respiratory arrest (CRA) | 2 (4.5%) | 1(3.6%) | 1 (6.2%) | |
| Syncope, ventricular fibrillation (FV) | 1 (2.3%) | 1 (3.6%) | 0 (0%) | |
| Death by cardiac heart failure (CHF) | 3 (6.8%) | 2 (7.2%) | 1 (6.2%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández Suárez, N.; Viadero Ubierna, M.T.; Garde Basas, J.; Onecha de la Fuente, M.E.; Amigo Lanza, M.T.; Martin Gorria, G.; Rivas Pérez, A.; Ruiz Guerrero, L.; González-Lamuño, D. Description of a Cohort with a New Truncating MYBPC3 Variant for Hypertrophic Cardiomyopathy in Northern Spain. Genes 2023, 14, 840. https://doi.org/10.3390/genes14040840
Fernández Suárez N, Viadero Ubierna MT, Garde Basas J, Onecha de la Fuente ME, Amigo Lanza MT, Martin Gorria G, Rivas Pérez A, Ruiz Guerrero L, González-Lamuño D. Description of a Cohort with a New Truncating MYBPC3 Variant for Hypertrophic Cardiomyopathy in Northern Spain. Genes. 2023; 14(4):840. https://doi.org/10.3390/genes14040840
Chicago/Turabian StyleFernández Suárez, Natalia, María Teresa Viadero Ubierna, Jesús Garde Basas, María Esther Onecha de la Fuente, María Teresa Amigo Lanza, Gonzalo Martin Gorria, Adrián Rivas Pérez, Luis Ruiz Guerrero, and Domingo González-Lamuño. 2023. "Description of a Cohort with a New Truncating MYBPC3 Variant for Hypertrophic Cardiomyopathy in Northern Spain" Genes 14, no. 4: 840. https://doi.org/10.3390/genes14040840
APA StyleFernández Suárez, N., Viadero Ubierna, M. T., Garde Basas, J., Onecha de la Fuente, M. E., Amigo Lanza, M. T., Martin Gorria, G., Rivas Pérez, A., Ruiz Guerrero, L., & González-Lamuño, D. (2023). Description of a Cohort with a New Truncating MYBPC3 Variant for Hypertrophic Cardiomyopathy in Northern Spain. Genes, 14(4), 840. https://doi.org/10.3390/genes14040840