

Chronic Stress Alters Hippocampal Renin-Angiotensin-Aldosterone System Component Expression in an Aged Rat Model of Wolfram Syndrome
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Treatment and Sample Collection
2.3. Sample Preparation and Gene Expression Analyses
2.4. Statistical Analysis
3. Results
3.1. Agtr1a, Agtr1b, Agtr2 and Bdkrb1 Levels Are Downregulated in the Hippocampi of WS Rats Receiving Chronic Treatment
3.2. RAAS Component Expression Was Unchanged in the Brain Stems of WS Rats Receiving Chronic Treatment
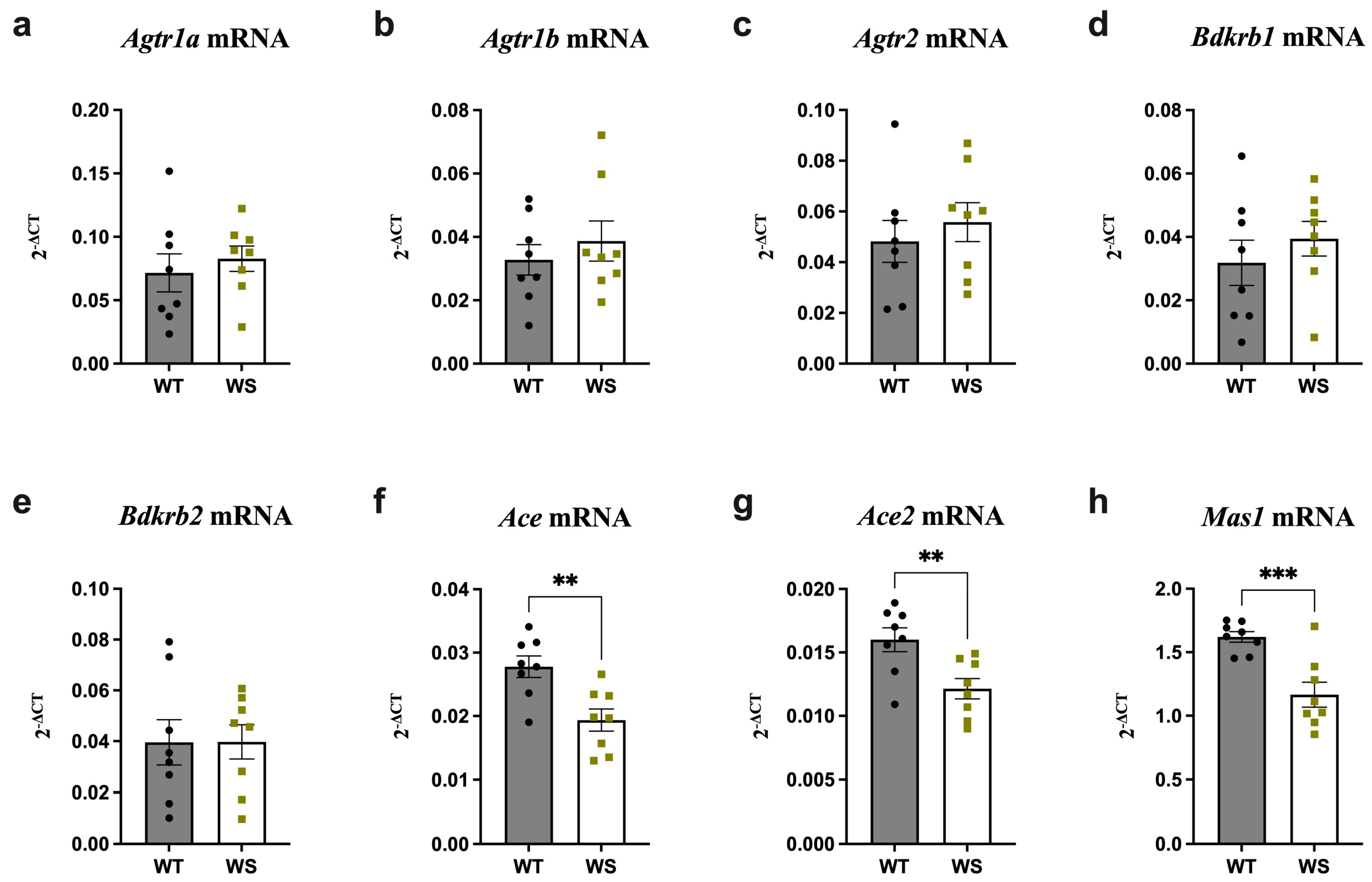
3.3. Ace, Ace2 and Mas1 Were Significantly Downregulated in the Hippocampi of Treatment-Naïve WS Rats
3.4. Ace Was Significantly Upregulated and Agtr2 Downregulated in the Brain Stems of Treatment-Naïve WS Rats
4. Discussion
5. Conclusions
6. Limitations of the Study
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
| 7,8-DHF | 7,8-dihydroxyflavone |
| Ace | Angiotensin I converting enzyme |
| Ace2 | Angiotensin I converting enzyme 2 |
| Agtr1a | Angiotensin II receptor type 1a |
| Agtr1b | Angiotensin II receptor type 1b |
| Agtr2 | Angiotensin II receptor type 2 |
| ARRIVE | Animal Research: Reporting of In Vivo Experiments |
| Bdkrb1 | Bradykinin receptor B1 |
| Bdkrb2 | Bradykinin receptor B2 |
| BDNF | Brain-derived neurotrophic factor |
| CNS | Central nervous system |
| DMSO | Dimethyl sulfoxide |
| ER | Endoplasmic reticulum |
| GABA | Gamma-aminobutyric acid |
| GLP-1R | Glucagon-like peptide 1 receptor |
| Hprt1 | Hypoxanthine-guanine phosphoribosyltransferase |
| LIR | Liraglutide |
| MAM | Mitochondria-associated ER membraane |
| Mas1 | MAS1 proto-oncogene, G protein-coupled receptor |
| PBS | Phosphate-buffered saline |
| PEG-300 | Polyethylene glycol-300 |
| RAAS | Renin-angiotensin-aldosterone system |
| SEM | Standard error of the mean |
| TrkB | Tropomyosin receptor kinase B |
| VEH | Vehicle |
| WFS1 | Wolframin/Wolfram Syndrome 1 |
| WS | Wolfram Syndrome |
| WT | Wild-type |
References
- Barrett, T.G.; Bundey, S.E. Wolfram (DIDMOAD) Syndrome. J. Med. Genet. 1997, 34, 838–841. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Tanizawa, Y.; Wasson, J.; Behn, P.; Kalidas, K.; Bernal-Mizrachi, E.; Mueckler, M.; Marshall, H.; Donis-Keller, H.; Crock, P.; et al. A Gene Encoding a Transmembrane Protein Is Mutated in Patients with Diabetes Mellitus and Optic Atrophy (Wolfram Syndrome). Nat. Genet. 1998, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.G.; Bundey, S.E.; Macleod, A.F. Neurodegeneration and Diabetes: UK Nationwide Study of Wolfram (DIDMOAD) Syndrome. Lancet 1995, 346, 1458–1463. [Google Scholar] [CrossRef]
- Medlej, R.; Wasson, J.; Baz, P.; Azar, S.; Salti, I.; Loiselet, J.; Permutt, A.; Halaby, G. Diabetes Mellitus and Optic Atrophy: A Study of Wolfram Syndrome in the Lebanese Population. J. Clin. Endocrinol. Metab. 2004, 89, 1656–1661. [Google Scholar] [CrossRef]
- Luuk, H.; Koks, S.; Plaas, M.; Hannibal, J.; Rehfeld, J.F.; Vasar, E. Distribution of Wfs1 Protein in the Central Nervous System of the Mouse and Its Relation to Clinical Symptoms of the Wolfram Syndrome. J. Comp. Neurol. 2008, 509, 642–660. [Google Scholar] [CrossRef]
- Hofmann, S.; Philbrook, C.; Gerbitz, K.-D.; Bauer, M.F. Wolfram Syndrome: Structural and Functional Analyses of Mutant and Wild-Type Wolframin, the WFS1 Gene Product. Hum. Mol. Genet. 2003, 12, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 21 February 2021).
- GeneCards—Human Genes|Gene Database|Gene Search. Available online: https://www.genecards.org/ (accessed on 21 February 2021).
- Fonseca, S.G.; Fukuma, M.; Lipson, K.L.; Nguyen, L.X.; Allen, J.R.; Oka, Y.; Urano, F. WFS1 is a Novel Component of the Unfolded Protein Response and Maintains Homeostasis of the Endoplasmic Reticulum in Pancreatic β-Cells. J. Biol. Chem. 2005, 280, 39609–39615. [Google Scholar] [CrossRef] [PubMed]
- Takei, D.; Ishihara, H.; Yamaguchi, S.; Yamada, T.; Tamura, A.; Katagiri, H.; Maruyama, Y.; Oka, Y. WFS1 Protein Modulates the Free Ca(2+) Concentration in the Endoplasmic Reticulum. FEBS Lett. 2006, 580, 5635–5640. [Google Scholar] [CrossRef]
- Cagalinec, M.; Liiv, M.; Hodurova, Z.; Hickey, M.A.; Vaarmann, A.; Mandel, M.; Zeb, A.; Choubey, V.; Kuum, M.; Safiulina, D.; et al. Role of Mitochondrial Dynamics in Neuronal Development: Mechanism for Wolfram Syndrome. PLoS Biol. 2016, 14, e1002511. [Google Scholar] [CrossRef]
- La Morgia, C.; Maresca, A.; Amore, G.; Gramegna, L.L.; Carbonelli, M.; Scimonelli, E.; Danese, A.; Patergnani, S.; Caporali, L.; Tagliavini, F.; et al. Calcium Mishandling in Absence of Primary Mitochondrial Dysfunction Drives Cellular Pathology in Wolfram Syndrome. Sci. Rep. 2020, 10, 4785. [Google Scholar] [CrossRef]
- Angebault, C.; Fauconnier, J.; Patergnani, S.; Rieusset, J.; Danese, A.; Affortit, C.A.; Jagodzinska, J.; Mégy, C.; Quiles, M.; Cazevieille, C.; et al. ER-Mitochondria Cross-Talk Is Regulated by the Ca2+ Sensor NCS1 and Is Impaired in Wolfram Syndrome. Sci. Signal. 2018, 11, eaaq1380. [Google Scholar] [CrossRef] [PubMed]
- Crouzier, L.; Danese, A.; Yasui, Y.; Richard, E.M.; Liévens, J.-C.; Patergnani, S.; Couly, S.; Diez, C.; Denus, M.; Cubedo, N.; et al. Activation of the Sigma-1 Receptor Chaperone Alleviates Symptoms of Wolfram Syndrome in Preclinical Models. Sci. Transl. Med. 2022, 14, eabh3763. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, C.; Ishigaki, S.; Oslowski, C.M.; Fonseca, S.G.; Kato, T.; Urano, F. Valproate, a Mood Stabilizer, Induces WFS1 Expression and Modulates Its Interaction with ER Stress Protein GRP94. PLoS ONE 2009, 4, e4134. [Google Scholar] [CrossRef]
- Batjargal, K.; Tajima, T.; Jimbo, E.F.; Yamagata, T. Effect of 4-Phenylbutyrate and Valproate on Dominant Mutations of WFS1 Gene in Wolfram Syndrome. J. Endocrinol. Investig. 2020, 43, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Terasmaa, A.; Soomets, U.; Oflijan, J.; Punapart, M.; Hansen, M.; Matto, V.; Ehrlich, K.; Must, A.; Kõks, S.; Vasar, E. Wfs1 Mutation Makes Mice Sensitive to Insulin-like Effect of Acute Valproic Acid and Resistant to Streptozocin. J. Physiol. Biochem. 2011, 67, 381–390. [Google Scholar] [CrossRef]
- Lu, S.; Kanekura, K.; Hara, T.; Mahadevan, J.; Spears, L.D.; Oslowski, C.M.; Martinez, R.; Yamazaki-Inoue, M.; Toyoda, M.; Neilson, A.; et al. A Calcium-Dependent Protease as a Potential Therapeutic Target for Wolfram Syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, E5292–E5301. [Google Scholar] [CrossRef]
- Abreu, D.; Stone, S.I.; Pearson, T.S.; Bucelli, R.C.; Simpson, A.N.; Hurst, S.; Brown, C.M.; Kries, K.; Onwumere, C.; Gu, H.; et al. A Phase Ib/IIa Clinical Trial of Dantrolene Sodium in Patients with Wolfram Syndrome. JCI Insight 2021, 6, e145188. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Li, Y.; Hu, R.; Gong, M.; Chai, M.; Ma, X.; Cha, J.; Guo, P.; Yang, K.; Li, M.; et al. Modeling Disrupted Synapse Formation in Wolfram Syndrome Using HESCs-Derived Neural Cells and Cerebral Organoids Identifies Riluzole as a Therapeutic Molecule. Mol. Psychiatry 2023, 1–14. [Google Scholar] [CrossRef]
- Mullard, A. Amylyx’s ALS Therapy Secures FDA Approval, as Regulatory Flexibility Trumps Underwhelming Data. Nat. Rev. Drug Discov. 2022, 21, 786. [Google Scholar] [CrossRef]
- Kitamura, R.A.; Maxwell, K.G.; Ye, W.; Kries, K.; Brown, C.M.; Augsornworawat, P.; Hirsch, Y.; Johansson, M.M.; Weiden, T.; Ekstein, J.; et al. Multidimensional Analysis and Therapeutic Development Using Patient IPSC–Derived Disease Models of Wolfram Syndrome. JCI Insight 2022, 7, e156549. [Google Scholar] [CrossRef]
- Rigoli, L.; Caruso, V.; Salzano, G.; Lombardo, F. Wolfram Syndrome 1: From Genetics to Therapy. Int. J. Env. Res. Public Health 2022, 19, 3225. [Google Scholar] [CrossRef] [PubMed]
- Toots, M.; Seppa, K.; Jagomäe, T.; Koppel, T.; Pallase, M.; Heinla, I.; Terasmaa, A.; Plaas, M.; Vasar, E. Preventive Treatment with Liraglutide Protects against Development of Glucose Intolerance in a Rat Model of Wolfram Syndrome. Sci. Rep. 2018, 8, 10183. [Google Scholar] [CrossRef] [PubMed]
- Seppa, K.; Toots, M.; Reimets, R.; Jagomäe, T.; Koppel, T.; Pallase, M.; Hasselholt, S.; Mikkelsen, M.K.; Randel Nyengaard, J.; Vasar, E.; et al. GLP-1 Receptor Agonist Liraglutide Has a Neuroprotective Effect on an Aged Rat Model of Wolfram Syndrome. Sci. Rep. 2019, 9, 15742. [Google Scholar] [CrossRef]
- Seppa, K.; Jagomäe, T.; Kukker, K.G.; Reimets, R.; Pastak, M.; Vasar, E.; Terasmaa, A.; Plaas, M. Liraglutide, 7,8-DHF and Their Co-Treatment Prevents Loss of Vision and Cognitive Decline in a Wolfram Syndrome Rat Model. Sci. Rep. 2021, 11, 2275. [Google Scholar] [CrossRef]
- Jagomäe, T.; Seppa, K.; Reimets, R.; Pastak, M.; Plaas, M.; Hickey, M.A.; Kukker, K.G.; Moons, L.; De Groef, L.; Vasar, E.; et al. Early Intervention and Lifelong Treatment with GLP1 Receptor Agonist Liraglutide in a Wolfram Syndrome Rat Model with an Emphasis on Visual Neurodegeneration, Sensorineural Hearing Loss and Diabetic Phenotype. Cells 2021, 10, 3193. [Google Scholar] [CrossRef] [PubMed]
- Sedman, T.; Rünkorg, K.; Krass, M.; Luuk, H.; Plaas, M.; Vasar, E.; Volke, V. Exenatide Is an Effective Antihyperglycaemic Agent in a Mouse Model of Wolfram Syndrome 1. J. Diabetes Res. 2016, 2016, 9239530. [Google Scholar] [CrossRef]
- Kondo, M.; Tanabe, K.; Amo-Shiinoki, K.; Hatanaka, M.; Morii, T.; Takahashi, H.; Seino, S.; Yamada, Y.; Tanizawa, Y. Activation of GLP-1 Receptor Signalling Alleviates Cellular Stresses and Improves β Cell Function in a Mouse Model of Wolfram Syndrome. Diabetologia 2018, 61, 2189–2201. [Google Scholar] [CrossRef]
- Scully, K.J.; Wolfsdorf, J.I. Efficacy of GLP-1 Agonist Therapy in Autosomal Dominant WFS1-Related Disorder: A Case Report. Horm. Res. Paediatr. 2020, 93, 409–414. [Google Scholar] [CrossRef]
- Frontino, G.; Raouf, T.; Canarutto, D.; Tirelli, E.; Di Tonno, R.; Rigamonti, A.; Cascavilla, M.L.; Baldoli, C.; Scotti, R.; Leocani, L.; et al. Case Report: Off-Label Liraglutide Use in Children With Wolfram Syndrome Type 1: Extensive Characterization of Four Patients. Front. Pediatr. 2021, 9, 755365. [Google Scholar] [CrossRef]
- Punapart, M.; Seppa, K.; Jagomäe, T.; Liiv, M.; Reimets, R.; Kirillov, S.; Kaasik, A.; Moons, L.; De Groef, L.; Terasmaa, A.; et al. The Expression of RAAS Key Receptors, Agtr2 and Bdkrb1, Is Downregulated at an Early Stage in a Rat Model of Wolfram Syndrome. Genes 2021, 12, 1717. [Google Scholar] [CrossRef]
- Romaní-Pérez, M.; Outeiriño-Iglesias, V.; Moya, C.M.; Santisteban, P.; González-Matías, L.C.; Vigo, E.; Mallo, F. Activation of the GLP-1 Receptor by Liraglutide Increases ACE2 Expression, Reversing Right Ventricle Hypertrophy, and Improving the Production of SP-A and SP-B in the Lungs of Type 1 Diabetes Rats. Endocrinology 2015, 156, 3559–3569. [Google Scholar] [CrossRef]
- Sedman, T.; Heinla, K.; Vasar, E.; Volke, V. Liraglutide Treatment May Affect Renin and Aldosterone Release. Horm. Metab. Res. 2017, 49, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Perini, M.V.; Dmello, R.S.; Nero, T.L.; Chand, A.L. Evaluating the Benefits of Renin-Angiotensin System Inhibitors as Cancer Treatments. Pharmacol. Ther. 2020, 211, 107527. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Oliveira, A.; Nogueira, A.I.; Pereira, R.M.; Boas, W.W.V.; Dos Santos, R.A.S.; Simões e Silva, A.C. The Renin-Angiotensin System and Diabetes: An Update. Vasc. Health Risk Manag. 2008, 4, 787–803. [Google Scholar] [PubMed]
- Labandeira-Garcia, J.L.; Rodríguez-Perez, A.I.; Garrido-Gil, P.; Rodriguez-Pallares, J.; Lanciego, J.L.; Guerra, M.J. Brain Renin-Angiotensin System and Microglial Polarization: Implications for Aging and Neurodegeneration. Front. Aging Neurosci. 2017, 9, 129. [Google Scholar] [CrossRef]
- Guimond, M.-O.; Gallo-Payet, N. The Angiotensin II Type 2 Receptor in Brain Functions: An Update. Int. J. Hypertens. 2012, 2012, 351758. [Google Scholar] [CrossRef]
- Wright, J.W.; Harding, J.W. The Brain Renin–Angiotensin System: A Diversity of Functions and Implications for CNS Diseases. Pflug. Arch. Eur. J. Physiol. 2013, 465, 133–151. [Google Scholar] [CrossRef]
- Leung, P.S.; Chappell, M.C. A Local Pancreatic Renin-Angiotensin System: Endocrine and Exocrine Roles. Int. J. Biochem. Cell Biol. 2003, 35, 838–846. [Google Scholar] [CrossRef]
- Cao, X.; Lu, X.-M.; Tuo, X.; Liu, J.-Y.; Zhang, Y.-C.; Song, L.-N.; Cheng, Z.-Q.; Yang, J.-K.; Xin, Z. Angiotensin-Converting Enzyme 2 Regulates Endoplasmic Reticulum Stress and Mitochondrial Function to Preserve Skeletal Muscle Lipid Metabolism. Lipids Health Dis. 2019, 18, 207. [Google Scholar] [CrossRef]
- Escobales, N.; Nuñez, R.E.; Javadov, S. Mitochondrial Angiotensin Receptors and Cardioprotective Pathways. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1426–H1438. [Google Scholar] [CrossRef]
- Valenzuela, R.; Costa-Besada, M.A.; Iglesias-Gonzalez, J.; Perez-Costas, E.; Villar-Cheda, B.; Garrido-Gil, P.; Melendez-Ferro, M.; Soto-Otero, R.; Lanciego, J.L.; Henrion, D.; et al. Mitochondrial Angiotensin Receptors in Dopaminergic Neurons. Role in Cell Protection and Aging-Related Vulnerability to Neurodegeneration. Cell Death Dis. 2016, 7, e2427. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pallares, J.; Rey, P.; Parga, J.A.; Muñoz, A.; Guerra, M.J.; Labandeira-Garcia, J.L. Brain Angiotensin Enhances Dopaminergic Cell Death via Microglial Activation and NADPH-Derived ROS. Neurobiol. Dis. 2008, 31, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Sunanda, T.; Ray, B.; Mahalakshmi, A.M.; Bhat, A.; Rashan, L.; Rungratanawanich, W.; Song, B.-J.; Essa, M.M.; Sakharkar, M.K.; Chidambaram, S.B. Mitochondria-Endoplasmic Reticulum Crosstalk in Parkinson’s Disease: The Role of Brain Renin Angiotensin System Components. Biomolecules 2021, 11, 1669. [Google Scholar] [CrossRef] [PubMed]
- Scolding, N.J.; Kellar-Wood, H.F.; Shaw, C.; Shneerson, J.M.; Antount, N. Wolfram Syndrome: Hereditary Diabetes Mellitus with Brainstem and Optic Atrophy. Ann. Neurol. 1996, 39, 352–360. [Google Scholar] [CrossRef]
- Hershey, T.; Lugar, H.M.; Shimony, J.S.; Rutlin, J.; Koller, J.M.; Perantie, D.C.; Paciorkowski, A.R.; Eisenstein, S.A.; Permutt, M.A. Early Brain Vulnerability in Wolfram Syndrome. PLoS ONE 2012, 7, e40604. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Becker, L.; Deck, J. Evidence of Widespread Axonal Pathology in Wolfram Syndrome. Acta Neuropathol. 1999, 98, 304–308. [Google Scholar] [CrossRef]
- Takeda, K.; Inoue, H.; Tanizawa, Y.; Matsuzaki, Y.; Oba, J.; Watanabe, Y.; Shinoda, K.; Oka, Y. WFS1 (Wolfram Syndrome 1) Gene Product: Predominant Subcellular Localization to Endoplasmic Reticulum in Cultured Cells and Neuronal Expression in Rat Brain. Hum. Mol. Genet. 2001, 10, 477–484. [Google Scholar] [CrossRef]
- Plaas, M.; Seppa, K.; Reimets, R.; Jagomäe, T.; Toots, M.; Koppel, T.; Vallisoo, T.; Nigul, M.; Heinla, I.; Meier, R.; et al. Wfs1- Deficient Rats Develop Primary Symptoms of Wolfram Syndrome: Insulin-Dependent Diabetes, Optic Nerve Atrophy and Medullary Degeneration. Sci. Rep. 2017, 7, 10220. [Google Scholar] [CrossRef]
- Munshani, S.; Ibrahim, E.Y.; Domenicano, I.; Ehrlich, B.E. The Impact of Mutations in Wolframin on Psychiatric Disorders. Front. Pediatr. 2021, 9, 718132. [Google Scholar] [CrossRef]
- Mohite, S.; Sanches, M.; Teixeira, A.L. Exploring the Evidence Implicating the Renin-Angiotensin System (RAS) in the Physiopathology of Mood Disorders. Protein Pept. Lett. 2017, 27, 449–455. [Google Scholar] [CrossRef]
- McClean, P.L.; Jalewa, J.; Hölscher, C. Prophylactic Liraglutide Treatment Prevents Amyloid Plaque Deposition, Chronic Inflammation and Memory Impairment in APP/PS1 Mice. Behav. Brain Res. 2015, 293, 96–106. [Google Scholar] [CrossRef]
- Liu, W.; Jalewa, J.; Sharma, M.; Li, G.; Li, L.; Hölscher, C. Neuroprotective Effects of Lixisenatide and Liraglutide in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson’s Disease. Neuroscience 2015, 303, 42–50. [Google Scholar] [CrossRef]
- Yang, X.; Qiang, Q.; Li, N.; Feng, P.; Wei, W.; Hölscher, C. Neuroprotective Mechanisms of Glucagon-Like Peptide-1-Based Therapies in Ischemic Stroke: An Update Based on Preclinical Research. Front. Neurol. 2022, 13, 844697. [Google Scholar] [CrossRef]
- Lucius, R.; Gallinat, S.; Rosenstiel, P.; Herdegen, T.; Sievers, J.; Unger, T. The Angiotensin II Type 2 (AT2) Receptor Promotes Axonal Regeneration in the Optic Nerve of Adult Rats. J. Exp. Med. 1998, 188, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Diniz, C.R.A.F.; Casarotto, P.C.; Fred, S.M.; Biojone, C.; Castrén, E.; Joca, S.R.L. Antidepressant-like Effect of Losartan Involves TRKB Transactivation from Angiotensin Receptor Type 2 (AGTR2) and Recruitment of FYN. Neuropharmacology 2018, 135, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Hofman, Z.; de Maat, S.; Hack, C.E.; Maas, C. Bradykinin: Inflammatory Product of the Coagulation System. Clin. Rev. Allergy Immunol. 2016, 51, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Ifuku, M.; Färber, K.; Okuno, Y.; Yamakawa, Y.; Miyamoto, T.; Nolte, C.; Merrino, V.F.; Kita, S.; Iwamoto, T.; Komuro, I.; et al. Bradykinin-Induced Microglial Migration Mediated by B1-Bradykinin Receptors Depends on Ca2+ Influx via Reverse-Mode Activity of the Na+/Ca2+ Exchanger. J. Neurosci. 2007, 27, 13065–13073. [Google Scholar] [CrossRef]
- Saavedra, J.M.; Benicky, J. Brain and Peripheral Angiotensin II Play a Major Role in Stress. Stress 2007, 10, 185–193. [Google Scholar] [CrossRef]
- Petek, B.; Villa-Lopez, M.; Loera-Valencia, R.; Gerenu, G.; Winblad, B.; Kramberger, M.G.; Ismail, M.-A.-M.; Eriksdotter, M.; Garcia-Ptacek, S. Connecting the Brain Cholesterol and Renin–Angiotensin Systems: Potential Role of Statins and RAS-Modifying Medications in Dementia. J. Intern. Med. 2018, 284, 620–642. [Google Scholar] [CrossRef]
- Hemming, M.L.; Selkoe, D.J. Amyloid β-Protein Is Degraded by Cellular Angiotensin-Converting Enzyme (ACE) and Elevated by an ACE Inhibitor. J. Biol. Chem. 2005, 280, 37644–37650. [Google Scholar] [CrossRef]
- Kehoe, P.G.; Wong, S.; Al Mulhim, N.; Palmer, L.E.; Miners, J.S. Angiotensin-Converting Enzyme 2 Is Reduced in Alzheimer’s Disease in Association with Increasing Amyloid-β and Tau Pathology. Alzheimers Res. 2016, 8, 50. [Google Scholar] [CrossRef]
- Singh, P.K.; Chen, Z.-L.; Ghosh, D.; Strickland, S.; Norris, E.H. Increased Plasma Bradykinin Level Is Associated with Cognitive Impairment in Alzheimer’s Patients. Neurobiol. Dis. 2020, 139, 104833. [Google Scholar] [CrossRef] [PubMed]
- AbdAlla, S.; el Hakim, A.; Abdelbaset, A.; Elfaramawy, Y.; Quitterer, U. Inhibition of ACE Retards Tau Hyperphosphorylation and Signs of Neuronal Degeneration in Aged Rats Subjected to Chronic Mild Stress. BioMed Res. Int. 2015, 2015, 917156. [Google Scholar] [CrossRef] [PubMed]
- Fouda, A.Y.; Fagan, S.C.; Ergul, A. Brain Vasculature and Cognition. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Hellner, K.; Walther, T.; Schubert, M.; Albrecht, D. Angiotensin-(1–7) Enhances LTP in the Hippocampus through the G-Protein-Coupled Receptor Mas. Mol. Cell. Neurosci. 2005, 29, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Delpech, J.-C.; Pathak, D.; Varghese, M.; Kalavai, S.V.; Hays, E.C.; Hof, P.R.; Johnson, W.E.; Ikezu, S.; Medalla, M.; Luebke, J.I.; et al. Wolframin-1—Expressing Neurons in the Entorhinal Cortex Propagate Tau to CA1 Neurons and Impair Hippocampal Memory in Mice. Sci. Transl. Med. 2021, 13, eabe8455. [Google Scholar] [CrossRef]
- Chen, S.; Acosta, D.; Li, L.; Liang, J.; Chang, Y.; Wang, C.; Fitzgerald, J.; Morrison, C.; Goulbourne, C.N.; Nakano, Y.; et al. Wolframin Is a Novel Regulator of Tau Pathology and Neurodegeneration. Acta Neuropathol. 2022, 143, 547–569. [Google Scholar] [CrossRef]
- Chen, S.; Venkaraman, L.; Liang, J.; Nakano, Y.; Villegas, N.E.H.; Brown, C.; Urano, F.; Koks, S.; Serrano, G.E.; Beach, T.G.; et al. Deficiency of WFS1 Increases Vulnerability to Pathological Tau in Vitro and in Vivo. Alzheimer’s Dement. 2020, 16, e042085. [Google Scholar] [CrossRef]
- BioGPS—Your Gene Portal System. Available online: http://biogps.org/#goto=welcome (accessed on 2 June 2022).
- Chen, J.; Xie, J.-J.; Shi, K.-S.; Gu, Y.-T.; Wu, C.-C.; Xuan, J.; Ren, Y.; Chen, L.; Wu, Y.-S.; Zhang, X.-L.; et al. Glucagon-like Peptide-1 Receptor Regulates Endoplasmic Reticulum Stress-Induced Apoptosis and the Associated Inflammatory Response in Chondrocytes and the Progression of Osteoarthritis in Rat. Cell Death Dis. 2018, 9, 212. [Google Scholar] [CrossRef]
- Nuamnaichati, N.; Mangmool, S.; Chattipakorn, N.; Parichatikanond, W. Stimulation of GLP-1 Receptor Inhibits Methylglyoxal-Induced Mitochondrial Dysfunctions in H9c2 Cardiomyoblasts: Potential Role of Epac/PI3K/Akt Pathway. Front. Pharmacol. 2020, 11, 805. [Google Scholar] [CrossRef]
- Rodrigues Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes-E-Silva, A.C. The Anti-Inflammatory Potential of ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Evidence from Basic and Clinical Research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Istas, G.; Höges, S.; Yakoub, M.; Hendgen-Cotta, U.; Rassaf, T.; Rodriguez-Mateos, A.; Hering, L.; Grandoch, M.; Mergia, E.; et al. Angiotensin-(1-7)-Induced Mas Receptor Activation Attenuates Atherosclerosis through a Nitric Oxide-Dependent Mechanism in ApolipoproteinE-KO Mice. Pflug. Arch. 2018, 470, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Cork, S.C.; Richards, J.E.; Holt, M.K.; Gribble, F.M.; Reimann, F.; Trapp, S. Distribution and Characterisation of Glucagon-like Peptide-1 Receptor Expressing Cells in the Mouse Brain. Mol. Metab. 2015, 4, 718–731. [Google Scholar] [CrossRef]
- Hamilton, A.; Hölscher, C. Receptors for the Incretin Glucagon-like Peptide-1 Are Expressed on Neurons in the Central Nervous System. Neuroreport 2009, 20, 1161–1166. [Google Scholar] [CrossRef]
- Lee, C.H.; Yan, B.; Yoo, K.-Y.; Choi, J.H.; Kwon, S.-H.; Her, S.; Sohn, Y.; Hwang, I.K.; Cho, J.H.; Kim, Y.-M.; et al. Ischemia-Induced Changes in Glucagon-like Peptide-1 Receptor and Neuroprotective Effect of Its Agonist, Exendin-4, in Experimental Transient Cerebral Ischemia. J. Neurosci. Res. 2011, 89, 1103–1113. [Google Scholar] [CrossRef]
- Korol, S.V.; Jin, Z.; Babateen, O.; Birnir, B. GLP-1 and Exendin-4 Transiently Enhance GABAA Receptor–Mediated Synaptic and Tonic Currents in Rat Hippocampal CA3 Pyramidal Neurons. Diabetes 2014, 64, 79–89. [Google Scholar] [CrossRef]
- Zhou, C.; Li, C.; Yu, H.-M.; Zhang, F.; Han, D.; Zhang, G.-Y. Neuroprotection of γ-Aminobutyric Acid Receptor Agonists via Enhancing Neuronal Nitric Oxide Synthase (Ser847) Phosphorylation through Increased Neuronal Nitric Oxide Synthase and PSD95 Interaction and Inhibited Protein Phosphatase Activity in Cerebral Ischemia. J. Neurosci. Res. 2008, 86, 2973–2983. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Punapart, M.; Reimets, R.; Seppa, K.; Kirillov, S.; Gaur, N.; Eskla, K.-L.; Jagomäe, T.; Vasar, E.; Plaas, M. Chronic Stress Alters Hippocampal Renin-Angiotensin-Aldosterone System Component Expression in an Aged Rat Model of Wolfram Syndrome. Genes 2023, 14, 827. https://doi.org/10.3390/genes14040827
Punapart M, Reimets R, Seppa K, Kirillov S, Gaur N, Eskla K-L, Jagomäe T, Vasar E, Plaas M. Chronic Stress Alters Hippocampal Renin-Angiotensin-Aldosterone System Component Expression in an Aged Rat Model of Wolfram Syndrome. Genes. 2023; 14(4):827. https://doi.org/10.3390/genes14040827
Chicago/Turabian StylePunapart, Marite, Riin Reimets, Kadri Seppa, Silvia Kirillov, Nayana Gaur, Kattri-Liis Eskla, Toomas Jagomäe, Eero Vasar, and Mario Plaas. 2023. "Chronic Stress Alters Hippocampal Renin-Angiotensin-Aldosterone System Component Expression in an Aged Rat Model of Wolfram Syndrome" Genes 14, no. 4: 827. https://doi.org/10.3390/genes14040827
APA StylePunapart, M., Reimets, R., Seppa, K., Kirillov, S., Gaur, N., Eskla, K.-L., Jagomäe, T., Vasar, E., & Plaas, M. (2023). Chronic Stress Alters Hippocampal Renin-Angiotensin-Aldosterone System Component Expression in an Aged Rat Model of Wolfram Syndrome. Genes, 14(4), 827. https://doi.org/10.3390/genes14040827