

Exome Analysis Reveals Novel Missense and Deletion Variants in the CC2D2A Gene as Causative of Joubert Syndrome

 , , , , and
, , , , and
Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Family Trio
2.2. Whole Exome Sequencing
2.3. Sanger Sequencing and Quantitative Polymerase Chain Reaction
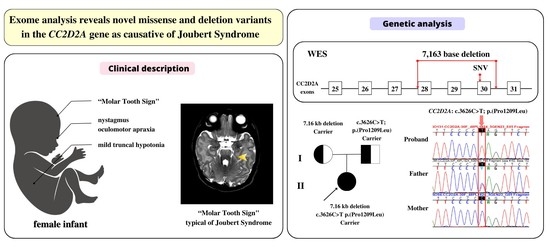
3. Results
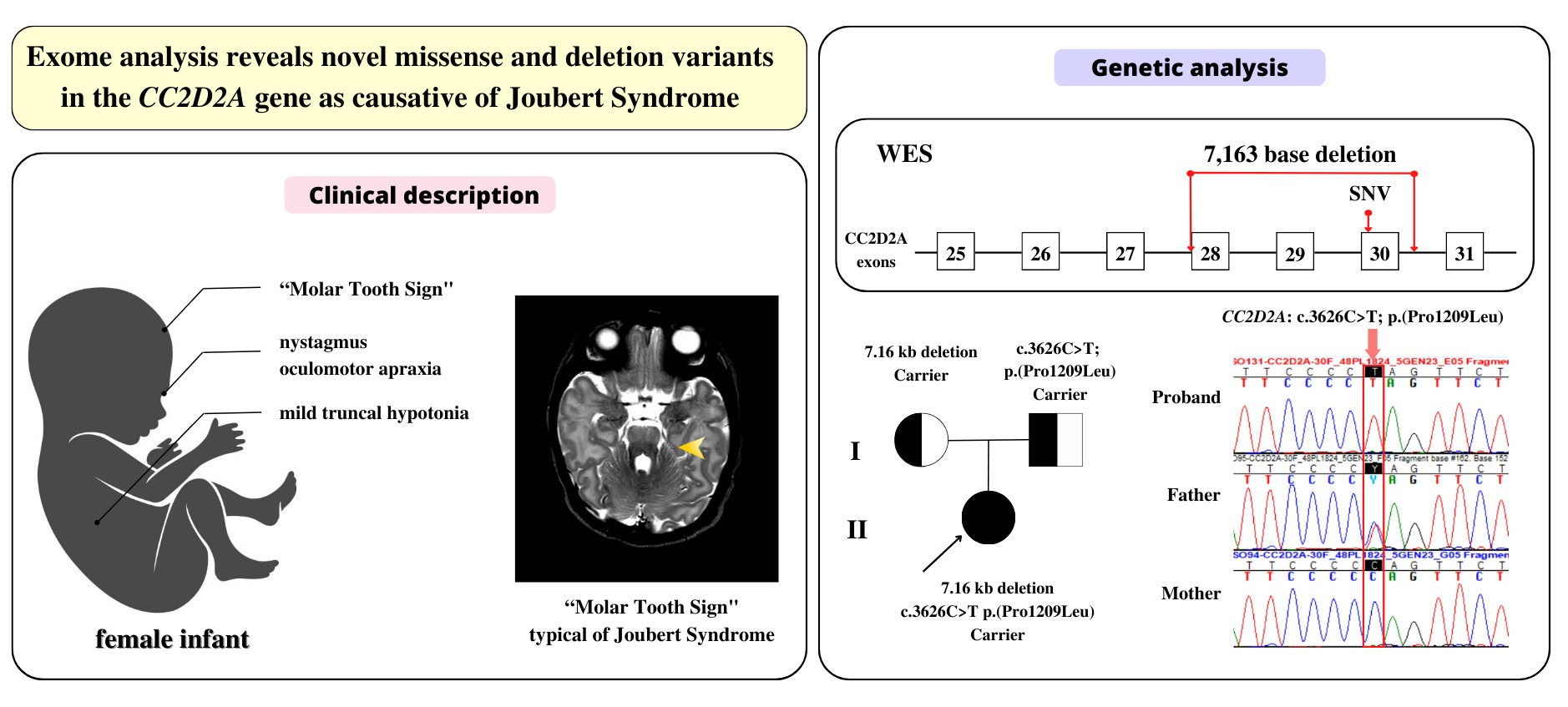
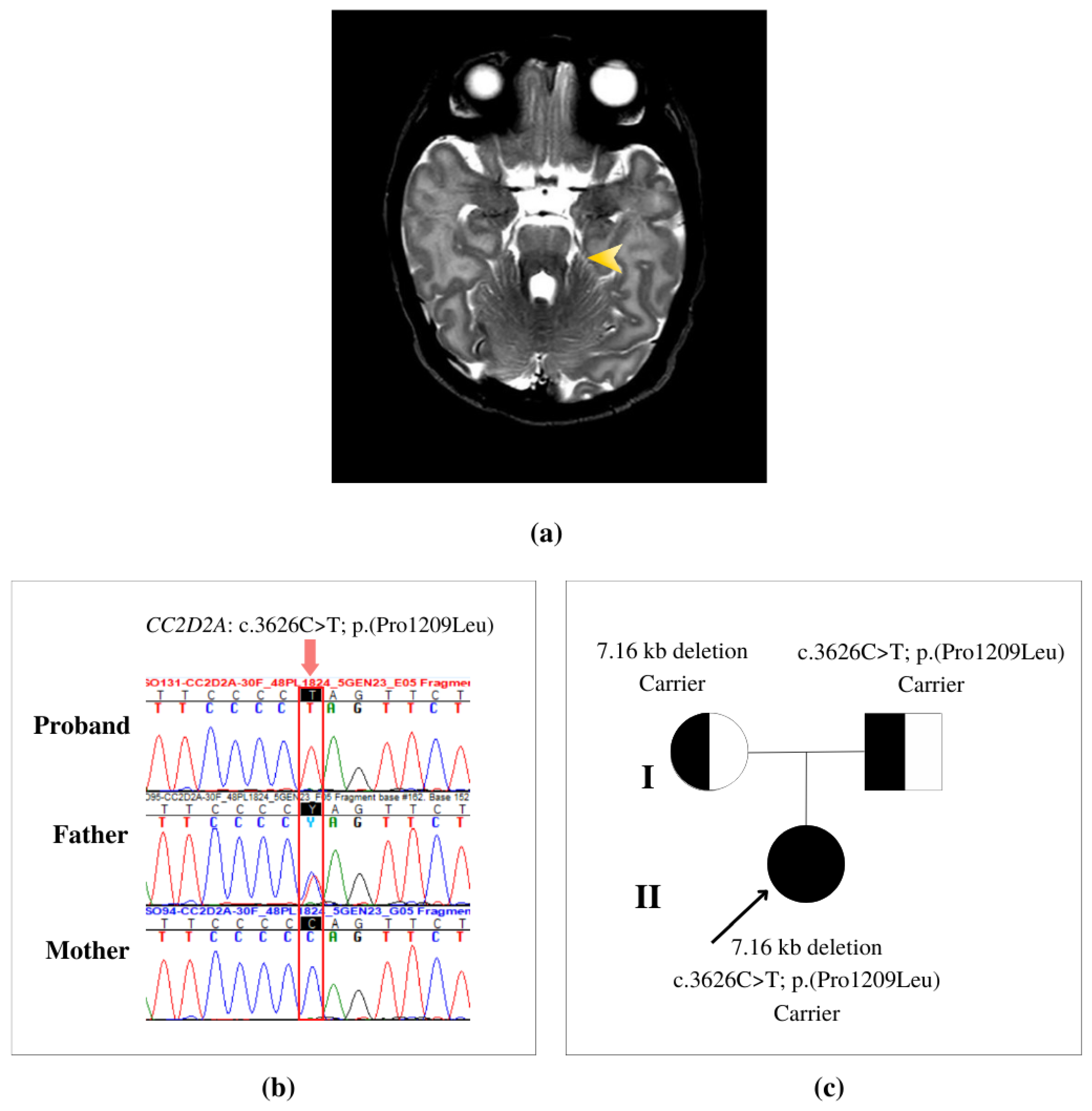
3.1. Clinical Description
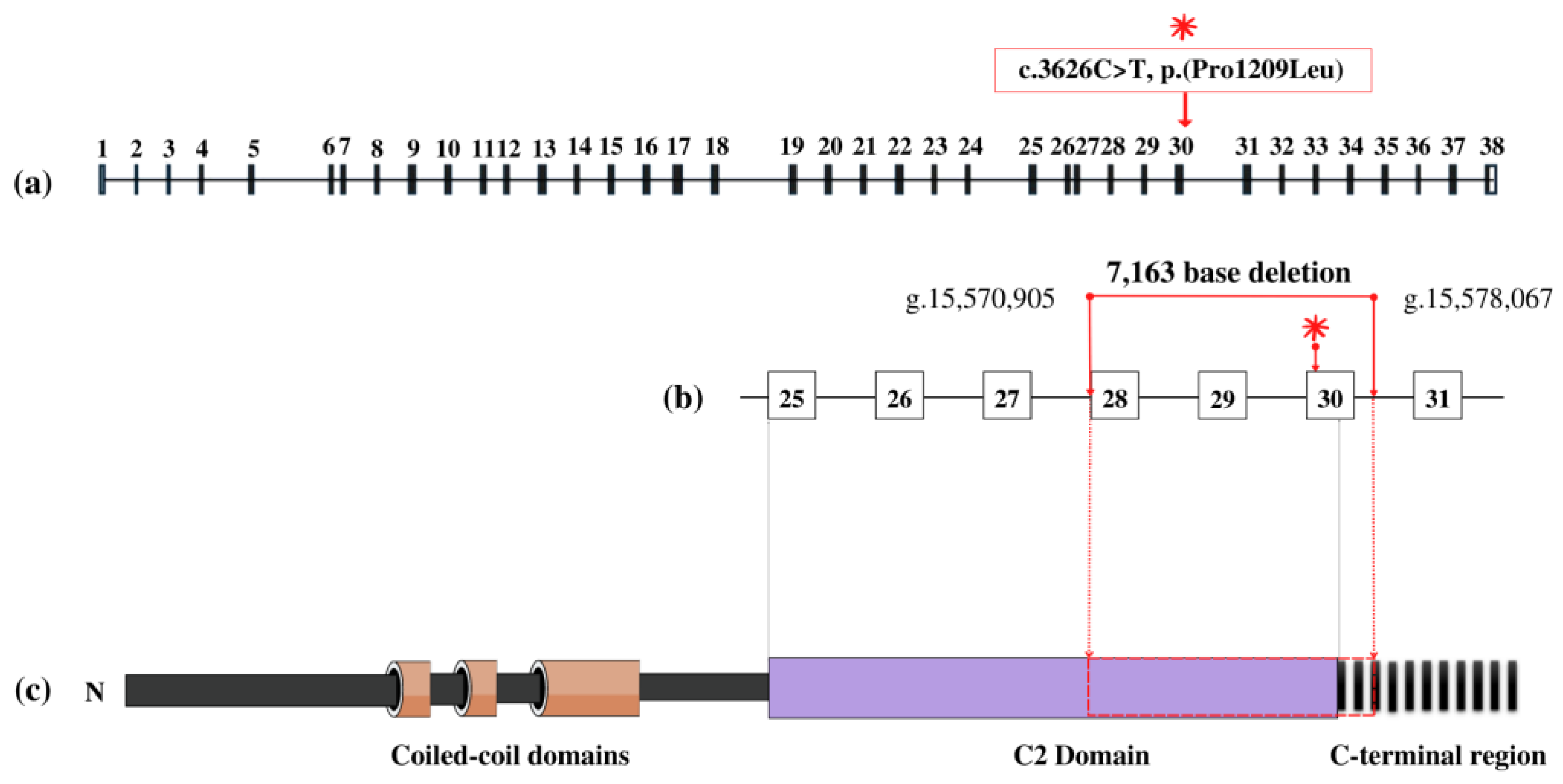
3.2. Genetic Findings
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harion, M.; Qebibo, L.; Riquet, A.; Rougeot, C.; Afenjar, A.; Garel, C.; Louha, M.; Lacaze, E.; Audic-Gérard, F.; Barth, M.; et al. New insights into CC2D2A-related Joubert syndrome. J. Med. Genet. 2022. published online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Parisi, M.A.; Doherty, D.; Chance, P.F.; Glass, I.A. Joubert syndrome (and related disorders) (OMIM 213300). Eur. J. Hum. Genet. 2007, 15, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Nuovo, S.; Bacigalupo, I.; Ginevrino, M.; Battini, R.; Bertini, E.; Borgatti, R.; Casella, A.; Micalizzi, A.; Nardella, M.; Romaniello, R.; et al. Age and sex prevalence estimate of Joubert syndrome in Italy. Neurology 2020, 94, e797–e801. [Google Scholar] [CrossRef] [PubMed]
- Kroes, H.Y.; Monroe, G.R.; van der Zwaag, B.; Duran, K.J.; de Kovel, C.G.; van Roosmalen, M.J.; Harakalova, M.; Nijman, I.J.; Kloosterman, W.P.; Giles, R.H.; et al. Joubert syndrome: Genotyping a Northern European patient cohort. Eur. J. Hum. Genet. 2016, 24, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Barroso-Gil, M.; Olinger, E.; Ramsbottom, S.A.; Molinari, E.; Miles, C.G.; Sayer, J.A. Update of genetic variants in CEP120 and CC2D2A-With an emphasis on genotype-phenotype correlations, tissue specific transcripts and exploring mutation specific exon skipping therapies. Mol. Genet. Genom. Med. 2021, 9, e1603. [Google Scholar] [CrossRef] [PubMed]
- Bachmann-Gagescu, R.; Dona, M.; Hetterscchijt, L.; Tonnaer, E.; Peters, T.; de Vrieze, E.; Mans, D.A.; van Beersum, S.E.; Phelps, I.G.; Arts, H.H.; et al. The ciliopathy protein CC2D2A associates with NINL and functions in Rab8-MICAL3-regulated vesicle trafficking. PLoS Genet. 2015, 11, e1005575. [Google Scholar] [CrossRef] [PubMed]
- Mougou-Zerelli, S.; Thomas, S.; Szenker, E.; Audollent, S.; Elkhartoufi, N.; Babarit, C.; Romano, S.; Salmon, R.; Amiel, J.; Esculpavit, C.; et al. CC2D2A mutations in Meckel and Joubert syndromes indicate a genotype–phenotype correlation. Hum. Mutat. 2009, 30, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Matthijs, G.; Souche, E.; Alders, M.; Corveleyn, A.; Eck, S.; Feenstra, I.; Race, V.; Sistermans, E.; Sturm, M.; Weiss, M.; et al. Guidelines for diagnostic next-generation sequencing. Eur. J. Hum. Genet. 2016, 24, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Gorden, N.T.; Arts, H.H.; Parisi, M.A.; Coene, K.L.M.; Letteboer, S.J.F.; van Beersum, S.E.C.; Mans, D.A.; Hikida, A.; Eckert, M.; Knutzen, D.; et al. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am. J. Hum. Genet. 2008, 83, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.X.; Lu, X.G.; Liu, J.X.; Xu, L.; Shang, N.; Guo, L.; OuYang, Y.C. Case report and a brief review: Analysis and challenges of prenatal imaging phenotypes and genotypes in Joubert syndrome. Front. Genet. 2022, 13, 1038274. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Xie, J.; Yu, S.; Luo, H.; Wu, W.; Xu, Z. Clinical and genetic analysis for a Joubert syndrome family with CC2D2A gene mutations. Zhonghua Er Ke Za Zhi = Chin. J. Pediatr. 2015, 53, 431–435. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabrita Pinto, R.L.; Viaggi, S.; Canale, E.; Martinez Popple, M.; Capra, V.; Conteduca, G.; Testa, B.; Coviello, D.; Covone, A.E. Exome Analysis Reveals Novel Missense and Deletion Variants in the CC2D2A Gene as Causative of Joubert Syndrome. Genes 2023, 14, 810. https://doi.org/10.3390/genes14040810
Cabrita Pinto RL, Viaggi S, Canale E, Martinez Popple M, Capra V, Conteduca G, Testa B, Coviello D, Covone AE. Exome Analysis Reveals Novel Missense and Deletion Variants in the CC2D2A Gene as Causative of Joubert Syndrome. Genes. 2023; 14(4):810. https://doi.org/10.3390/genes14040810
Chicago/Turabian StyleCabrita Pinto, Rute Luísa, Silvia Viaggi, Edoardo Canale, Marina Martinez Popple, Valeria Capra, Giuseppina Conteduca, Barbara Testa, Domenico Coviello, and Angela Elvira Covone. 2023. "Exome Analysis Reveals Novel Missense and Deletion Variants in the CC2D2A Gene as Causative of Joubert Syndrome" Genes 14, no. 4: 810. https://doi.org/10.3390/genes14040810
APA StyleCabrita Pinto, R. L., Viaggi, S., Canale, E., Martinez Popple, M., Capra, V., Conteduca, G., Testa, B., Coviello, D., & Covone, A. E. (2023). Exome Analysis Reveals Novel Missense and Deletion Variants in the CC2D2A Gene as Causative of Joubert Syndrome. Genes, 14(4), 810. https://doi.org/10.3390/genes14040810