
CRISPR/Cas9 Gene Editing System Can Alter Gene Expression and Induce DNA Damage Accumulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. Cell Culture and Transfection
2.3. Detection of Gene Editing Efficiency via T7EI Assay
2.4. Transcriptome Sequencing and Analysis
2.5. CCK-8 Assay
2.6. Western Blot Analysis
3. Results
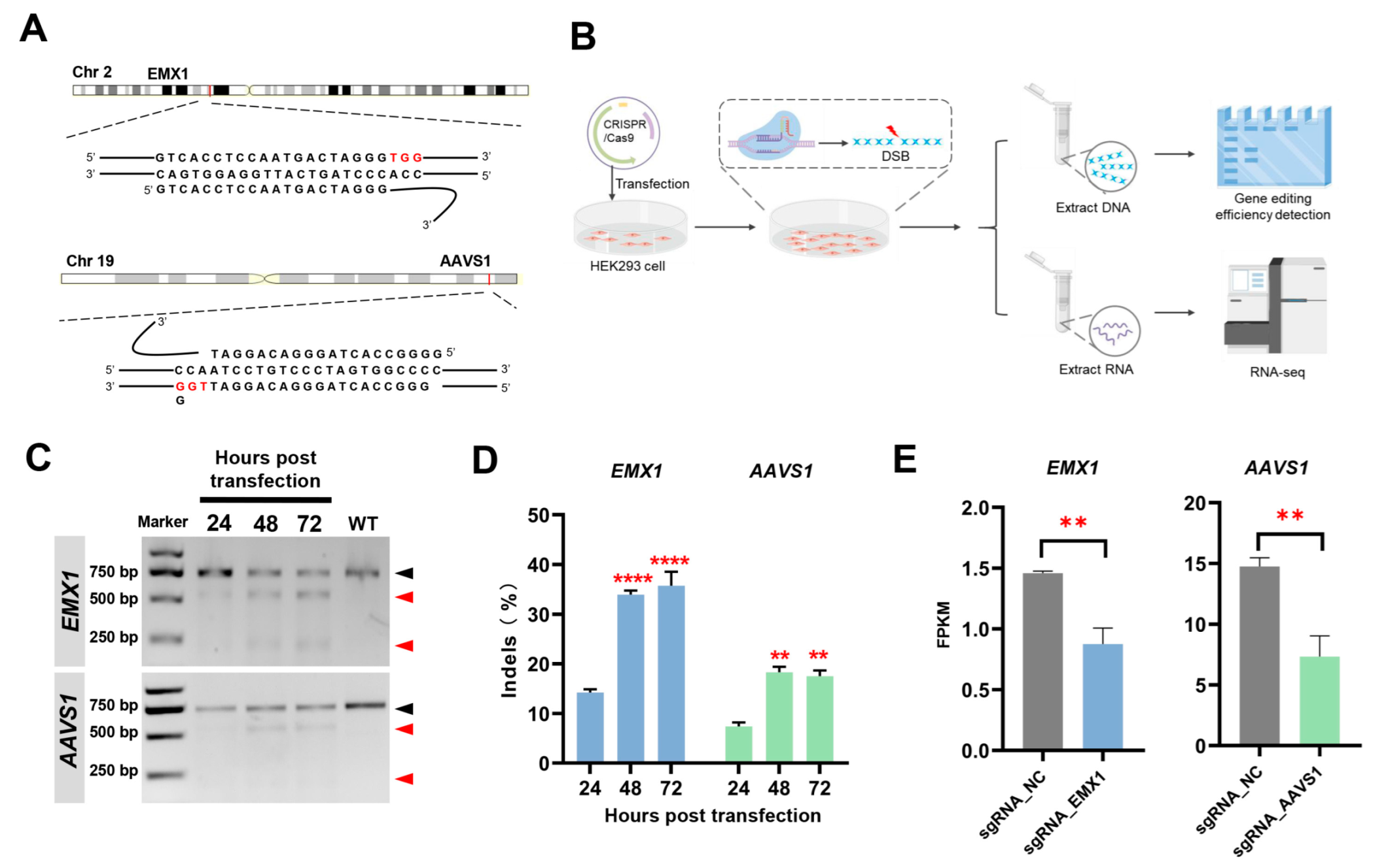
3.1. Experimental Design
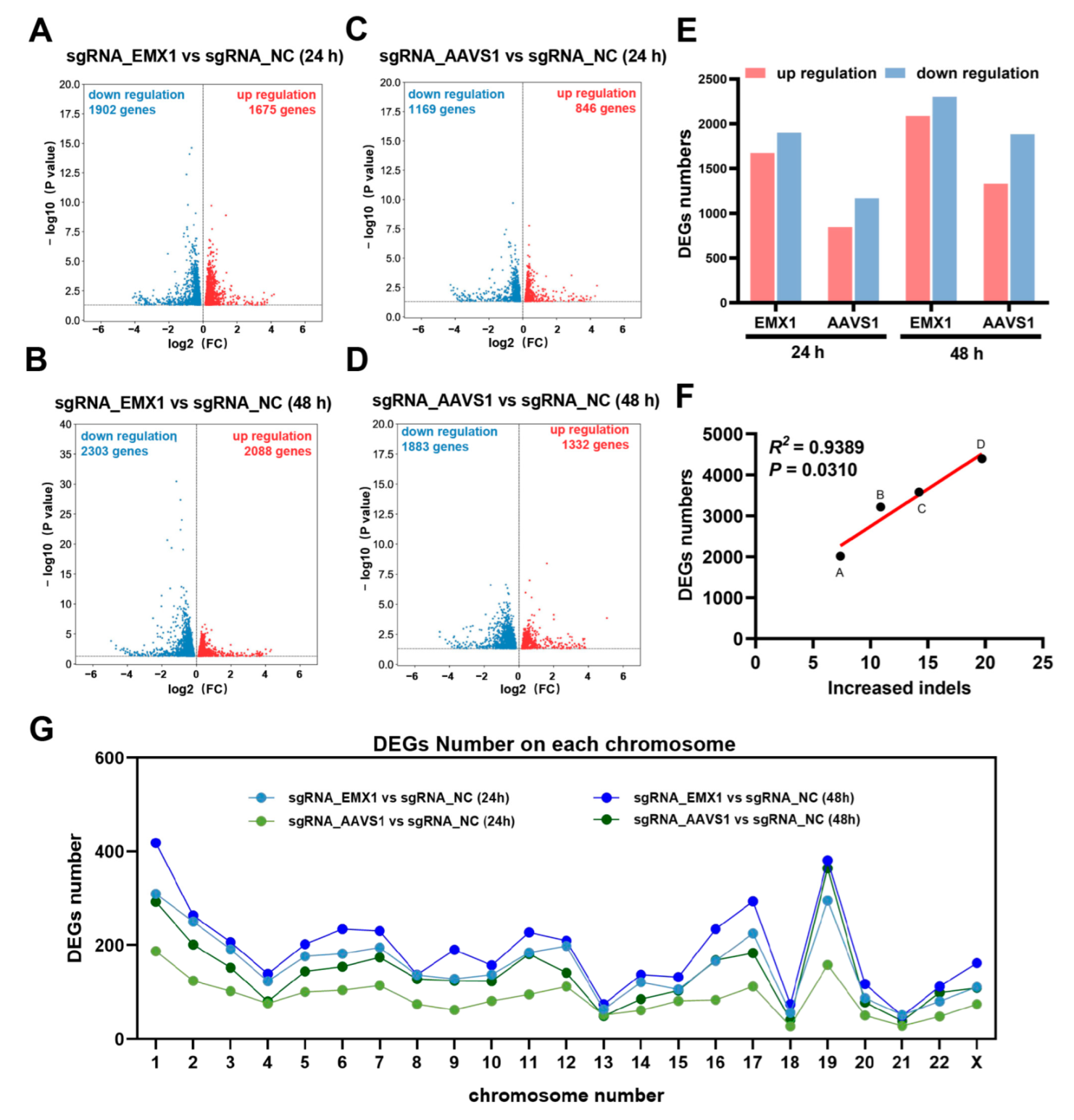
3.2. Transcriptomic Analysis Revealed that Gene Editing Reshapes Gene Expression
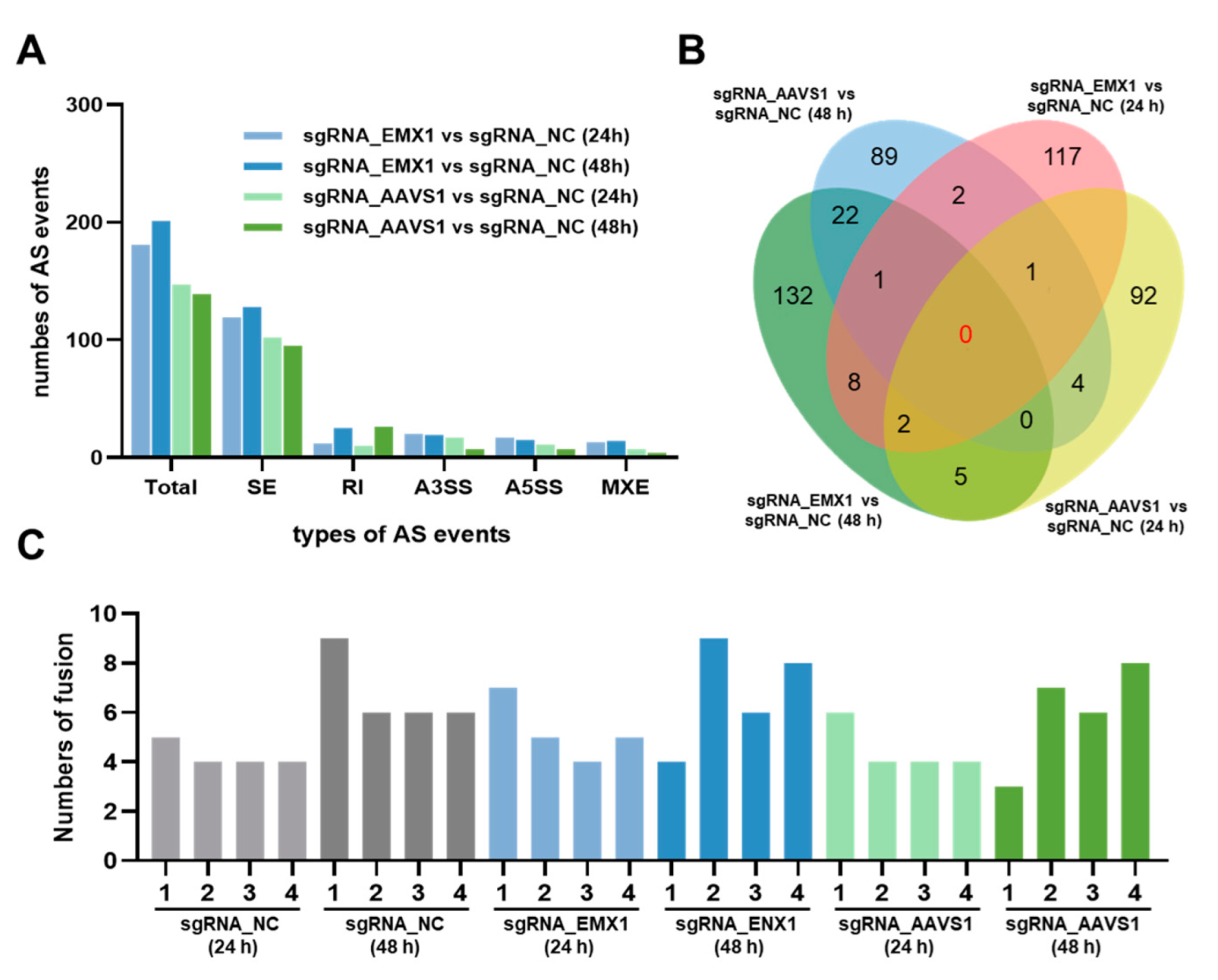
3.3. Gene Editing Randomly Induces Alternative Splicing and Little Fusion Genes
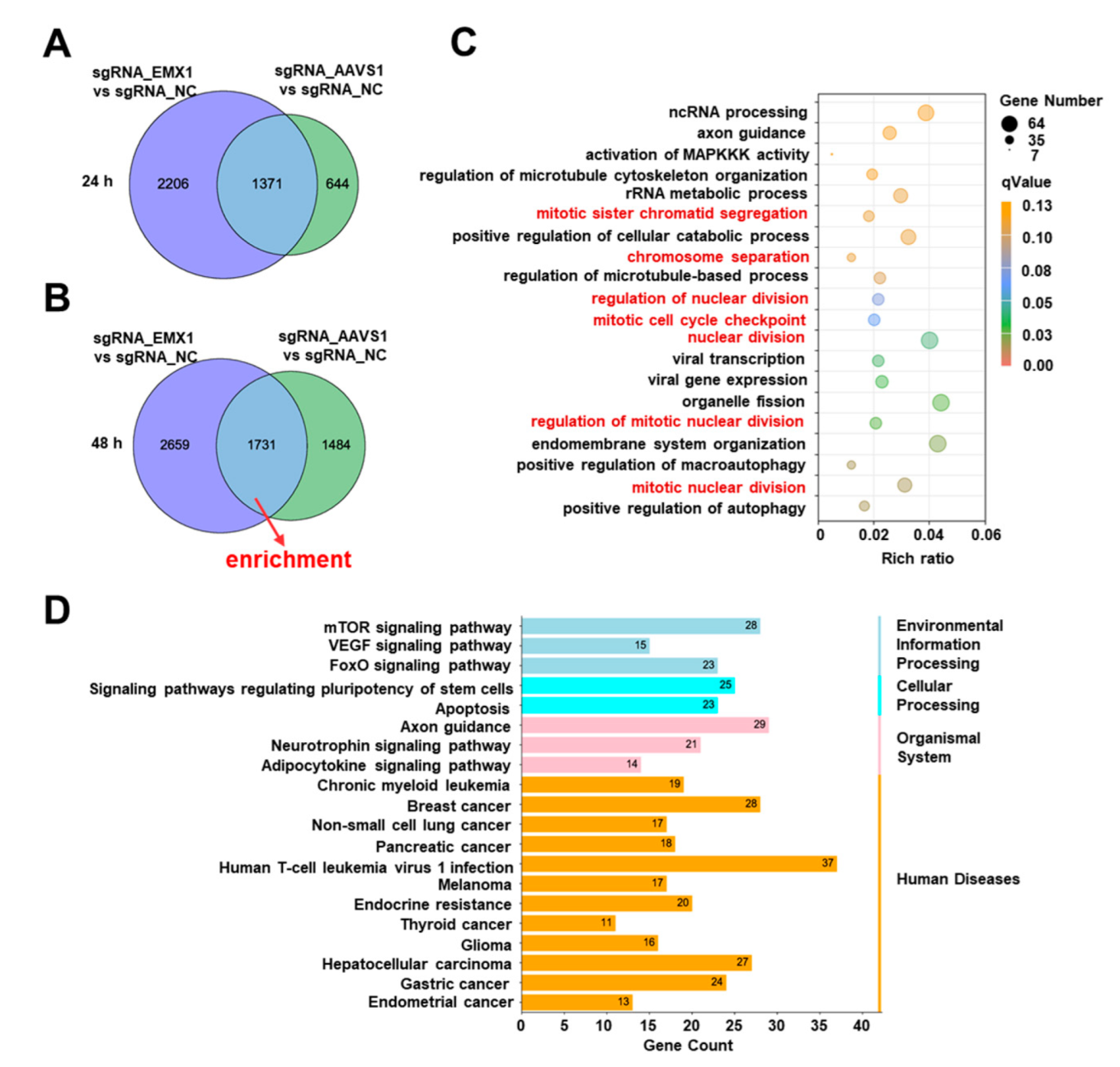
3.4. Gene Editing Altered Multiple Fundamental Biological Processes and Pathways Associated with Cancer and Other Diseases
3.5. CRISPR Gene Editing Activates DNA Damage Markers γH2AX
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef]
- Sapranauskas, R.; Gasiunas, G.; Fremaux, C.; Barrangou, R.; Horvath, P.; Siksnys, V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011, 39, 9275–9282. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. eLife 2013, 2, e00471. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Cullot, G.; Boutin, J.; Toutain, J.; Prat, F.; Pennamen, P.; Rooryck, C.; Teichmann, M.; Rousseau, E.; Lamrissi-Garcia, I.; Guyonnet-Duperat, V.; et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 2019, 10, 1136. [Google Scholar] [CrossRef] [PubMed]
- Boutin, J.; Rosier, J.; Cappellen, D.; Prat, F.; Toutain, J.; Pennamen, P.; Bouron, J.; Rooryck, C.; Merlio, J.P.; Lamrissi-Garcia, I.; et al. CRISPR-Cas9 globin editing can induce megabase-scale copy-neutral losses of heterozygosity in hematopoietic cells. Nat. Commun. 2021, 12, 4922. [Google Scholar] [CrossRef]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Sun, L.; Yao, Y.; Zhang, C.-Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat. Genet. 2021, 53, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Enache, O.M.; Rendo, V.; Abdusamad, M.; Lam, D.; Davison, D.; Pal, S.; Currimjee, N.; Hess, J.; Pantel, S.; Nag, A.; et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020, 52, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Wang, Q.; Mendez-Dorantes, C.; Burns, K.H.; Chiarle, R. Frequency and mechanisms of LINE-1 retrotransposon insertions at CRISPR/Cas9 sites. Nat. Commun. 2022, 13, 3685. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Sharma, A.R.; Bhattacharya, M.; Lee, S.-S.; Chakraborty, C. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol. Ther. 2021, 29, 571–586. [Google Scholar] [CrossRef]
- Nahmad, A.D.; Reuveni, E.; Goldschmidt, E.; Tenne, T.; Liberman, M.; Horovitz-Fried, M.; Khosravi, R.; Kobo, H.; Reinstein, E.; Madi, A.; et al. Frequent aneuploidy in primary human T cells after CRISPR–Cas9 cleavage. Nat. Biotechnol. 2022, 40, 1807–1813. [Google Scholar] [CrossRef]
- Wu, J.; Zou, Z.; Liu, Y.; Liu, X.; Zhangding, Z.; Xu, M.; Hu, J. CRISPR/Cas9-induced structural variations expand in T lymphocytes in vivo. Nucleic Acids Res. 2022, 50, 11128–11137. [Google Scholar] [CrossRef] [PubMed]
- Höijer, I.; Emmanouilidou, A.; Östlund, R.; van Schendel, R.; Bozorgpana, S.; Tijsterman, M.; Feuk, L.; Gyllensten, U.; Hoed, M.D.; Ameur, A. CRISPR-Cas9 induces large structural variants at on-target and off-target sites in vivo that segregate across generations. Nat. Commun. 2022, 13, 627. [Google Scholar] [CrossRef]
- Ottaviano, G.; Georgiadis, C.; Gkazi, S.A.; Syed, F.; Zhan, H.; Etuk, A.; Preece, R.; Chu, J.; Kubat, A.; Adams, S.; et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci. Transl. Med. 2022, 14, eabq3010. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and beta-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, G.; Gao, Z.; Li, J.; Wang, J.; Zhu, X.; Ma, R.; Yang, J.; Zhou, Y.; Hu, K.; et al. Global chromosome rearrangement induced by CRISPR-Cas9 reshapes the genome and transcriptome of human cells. Nucleic Acids Res. 2022, 50, 3456–3474. [Google Scholar] [CrossRef]
- Zhang, Q.; Fu, Y.; Thakur, C.; Bi, Z.; Wadgaonkar, P.; Qiu, Y.; Xu, L.; Rice, M.; Zhang, W.; Almutairy, B.; et al. CRISPR-Cas9 gene editing causes alternative splicing of the targeting mRNA. Biochem. Biophys. Res. Commun. 2020, 528, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yin, J.; Zhang-Ding, Z.; Xin, C.; Liu, M.; Wang, Y.; Ai, C.; Hu, J. In-depth assessment of the PAM compatibility and editing activities of Cas9 variants. Nucleic Acids Res. 2021, 49, 8785–8795. [Google Scholar] [CrossRef]
- Sinha, S.; Barbosa, K.; Cheng, K.; Leiserson, M.D.M.; Jain, P.; Deshpande, A.; Wilson, D.M., III.; Ryan, B.M.; Luo, J.; Ronai, Z.A.; et al. A systematic genome-wide mapping of oncogenic mutation selection during CRISPR-Cas9 genome editing. Nat. Commun. 2021, 12, 6512. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Farhan, M.; Wang, H.; Gaur, U.; Little, P.; Xu, J.; Zheng, W. FOXO Signaling Pathways as Therapeutic Targets in Cancer. Int. J. Biol. Sci. 2017, 13, 815–827. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Li, H.; Han, Y.; Song, Y.; Wei, M.; Fang, M.; Sun, Y. CRISPR/Cas9 Gene Editing System Can Alter Gene Expression and Induce DNA Damage Accumulation. Genes 2023, 14, 806. https://doi.org/10.3390/genes14040806
Yang L, Li H, Han Y, Song Y, Wei M, Fang M, Sun Y. CRISPR/Cas9 Gene Editing System Can Alter Gene Expression and Induce DNA Damage Accumulation. Genes. 2023; 14(4):806. https://doi.org/10.3390/genes14040806
Chicago/Turabian StyleYang, Lan, Hao Li, Yao Han, Yingjie Song, Mingchen Wei, Mengya Fang, and Yansong Sun. 2023. "CRISPR/Cas9 Gene Editing System Can Alter Gene Expression and Induce DNA Damage Accumulation" Genes 14, no. 4: 806. https://doi.org/10.3390/genes14040806
APA StyleYang, L., Li, H., Han, Y., Song, Y., Wei, M., Fang, M., & Sun, Y. (2023). CRISPR/Cas9 Gene Editing System Can Alter Gene Expression and Induce DNA Damage Accumulation. Genes, 14(4), 806. https://doi.org/10.3390/genes14040806