Abstract
The mustache toads Leptobrachium boringii and Leptobrachium liui are two attractive species in Megophryidae, in which adult males have mustache-like keratinized nuptial spines on their upper lip. However, both are under threat due to multiple factors, of which scientific studies are still very limited. In this study, two new complete mitochondrial genomes of L. boringii and L. liui were sequenced, assembled, and annotated based on next-generation sequencing. The mitogenome lengths of L. boringii and L. liui were found to be 17,100 and 17,501 bp, respectively, with both containing 13 protein coding genes, 23 tRNAs, 2 rRNAs, and 1 non-coding control region. Nucleotide diversity analyses indicate that atp8, atp6, and nad2 showed higher nucleotide diversity than cox1, cox3, and cytb. The intraspecific genetic distances among three different populations of L. boringii exceed 4%, and those between two populations of L. liui reach 7%. Phylogenetic relationships support their division into two subfamilies of Megophryidae (Leptobrachiinae and Megophryinae) as well as two species groups within Leptobrachium, corresponding to the number of keratinized nuptial spines (10–48 in the L. boringii species group vs. 2–6 in the L. liui species group). The two new mitogenomes reported in this study provide valuable data for future molecular evolutionary and conservation studies of the genus Leptobrachium and other Megophryidae toads.
1. Introduction
The toads of the family Megophryidae (Bonaparte, 1850) represent a group endemic to Asia and are found mainly distributed in tropical and subtropical forestry regions, ranging from northeast India, east Himalayas, eastward to South China, and southward to Southeast Asia [1]. For a long time, Megophryidae was known as Megophryinae, a subfamily within Pelobatidae (Bonaparte, 1850), until morphological studies showed that Megophryinae should be excluded from Pelobatidae based on skeletal structures; thus, it was suggested it be promoted to family level [2,3]. The validity and monophyly of Megophryidae has been supported by subsequent molecular phylogenetic studies [4,5]. At present, Megophryidae is widely recognized as an independent family that includes two subfamilies, the Leptobrachiinae and the Megophryinae, which comprise 179 and 129 species, respectively [6].
According to AmphibiaWeb (2023), Leptobrachium (Tschudi, 1838), the type genus of the subfamily Leptobrachiinae, includes 38 species of which 11 have been recorded in China, with 9 being endemic [7]. The toads in Leptobrachium prefer montane habitats in broad-leaf temperate forests at elevations of 700~1700 m, and they usually require slow-flowing streams with dense vegetation for breeding and forested terrestrial uplands to occupy outside the breeding season [7,8,9]. The taxonomy within the genus Leptobrachium is, however, debatable to some extent. It was originally divided into two subgenera, Vibrissaphora and Leptobrachium, according to whether or not adult males have keratinized nuptial spines [10,11]. There were also studies that further treated the spines in adult males as a basis for distinguishing Vibrissaphora and Leptobrachium as two distinct genera [7,12]. However, recent phylogenetic studies have proposed a single genus, Leptobrachium, including all species without subgeneric division, for the reason that Vibrissaphora species were not recovered as a single monophylum but dispersed within the lineage of Leptobrachium [8,13,14]. Leptobrachium species have a pair of large dark eyes with the upper half of the iris extraordinarily colored, such as sky-blue or green. Since these toads always hide in the daytime and come out at night, a study on their retina showed that the cell organization is principally adapted to enable sight at low light intensities [15]. This characteristic also seems to be useful for species identification and may bear phylogenetic signals; however, more data support for this speculation is still required [14]. Another unique aspect of the genus is its choice of cold season for breeding, whereas most frogs and toads breed in warmer months [16].
Leptobrachium boringii (Liu, 1945), known as the Emei mustache toad, is an attractive species in the genus. It was named after the mustache the males use as weapon for fighting during the breeding seasons, which is actually small keratinized nuptial spines growing on the upper lip [17]. The males are aggressive toward each other and outgrow the females to also aid in male–male combat, which is pretty rare in anurans [18]. Studies on reproductive behavior have indicated that males provide paternal care and larger males have higher mating success [19,20]. Both male combat and paternal care have been suggested to have adaptive significance in competing for nests, improving mating opportunities, and offspring survival [18,19,21]. Adult males of another species, L. liui (Pope, 1947), also have keratinized spines (mustache) on the upper lip but less in number than L. boringii (2–4 vs. 10–16) [22]. The distribution ranges of L. liui and L. boringii are basically separate but overlap at a few boundary areas (Figure 1). Leptobrachium liui is known as having two subspecies, L. liui liui and L. liui yaoshanensis, with two and four keratinized spines, respectively [22].
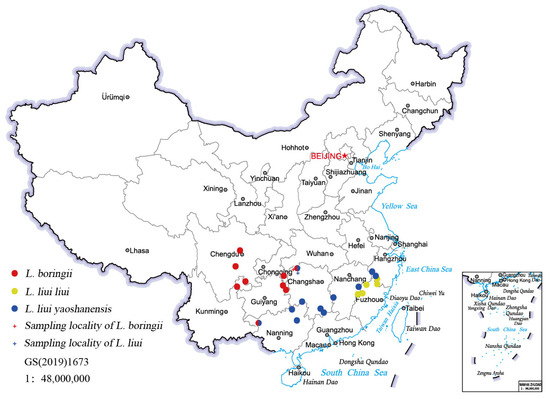
Figure 1.
The recorded distribution sites of L. boringii and L. liui (data from [7]).
Although most species in the Leptobrachium genus are only found very narrowly distributed in a few regions, L. boringii and L. liui are two exceptions that are found over a relatively large area. The current known populations of L. boringii are in Chongqing, Guangxi, Guizhou, Hunan, Yunnan, and Sichuan Provinces, and populations of L. liui are recorded in the Fujian, Zhejiang, Jiangxi, Hunan, Guangxi, and Guangdong Provinces of China (Figure 1; [7]). Although having relatively wide distribution, many populations of the two species are undergoing rapid decline due to over-catching, habitat loss, and degradation, among other factors [7,9]. The tadpoles of both L. boringii and L. liui take about four months to hatch and usually require three years to complete metamorphosis, which aggravates their chances of survival [22,23]. At present, L. boringii is classified as “Endangered” in the IUCN Red List of Threatened Species [24] and listed in the second class of National Key Protected Wild Animals of China [25], while L. liui was classified as “Least Concern” on the IUCN Red List of Threatened Species [26].
Speciation of amphibians has been driven by environmental factors and physiological adaptations during their short-term life cycle and long-term evolutionary history, which have shaped their current distribution and diversity patterns, showing similar characteristics or unique traits in morphology [22]. With more than 300 species in Megophryidae, a robust phylogenetic relationship reconstructed using mitogenomes would provide a basic framework to understand the phylogeny and trait evolution of this group. However, the available mitogenome data are still very limited. For instance, only 18 complete and 9 nearly complete mitogenomes of Megophryidae are available from NCBI to date. As for L. boringii, although it is relatively widely distributed, details of its population diversity and genetic differentiation are still largely unknown. There are two previous studies reporting the mitogenomes of L. boringii from two isolated populations: the Emei Mountains (EM) in Sichuan Province and Pengshui County (PS) in Chongqing Province [27,28]; however, these two mitogenomes were obtained using a traditional PCR-targeted sequencing method, and the sequences were incomplete. When it comes to L. liui, there was also a mitogenome reported from the population of the Jiulongshan National Nature Reserve (JLS) in Zhejiang Province. Similarly to L. boringii, however, it was only briefly described in the paper [29]. In this study, using next-generation sequencing (NGS) technology, we report two newly obtained complete mitogenomes of the genus Leptobrachium, one from L. boringii based on its easternmost population from the National Nature Reserve of Badagongshan in Sangzhi County (SZ) and the other from L. liui based on its northernmost population from the Zhangjiajie National Forest Park in Wulingyuan District (WLY). Both Sangzhi County and Wulingyuan District belong to Zhangjiajie City in Hunan Province and, interestingly, represent a boundary area of L. boringii and L. liu here (Figure 1). This study aims to: (1) analyze and describe the characteristics of the updated complete mitogenomes of L. boringii and L. liui in detail, (2) compare the genetic distances among the three representative populations of L. boringii and the two populations of L. liui, and (3) reconstruct the phylogenetic relationships of Megophryidae and present implications of this evolutionarily interesting group.
2. Materials and Methods
2.1. Sample Collection and Sequencing
The sample of L. boringii was collected in May 2021 from National Nature Reserve of Badagongshan, Sangzhi County, and the sample of L. liui was collected in September 2021 from Zhangjiajie National Forest Park, Wulingyuan District, both in Zhangjiajie City in Hunan Province, China. Permissions of the field survey for scientific purposes were approved by the local administration, and the collection of toads used in this study complied with the Wildlife Protection Law of China. According to the “3R principle” (Reduction, Replacement, and Refinement) of animal sampling, only one sample of each species was used in this study. All procedures of animal collection and treatment comply with the guidance of the Code of Practice for the Housing and Care of Animals. The specimens were euthanized and preserved in 95% alcohol as reference specimens, with a small liver sample used for molecular analysis. DNA extraction was conducted using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany), and a DNA library was then constructed using the VAHTS Universal DNA Library Prep Kit for Illumina V3 (Vazyme, Nanjing, China). High-throughput sequencing was performed in paired-end mode on the DNBSEQ-T7 platform (Complete Genomics and MGI Tech, Shenzhen, China), generating approximately 30 Gb of raw reads of 150 bp read length.
2.2. Sequence Assembly, Annotation, and Analysis
The complete mitogenomes of L. boringii and L. liui were assembled by NOVOPlasty 4.3 [30] based on the raw reads produced through NGS, both using the cytb gene (1141 bp) as seed sequences, from the previous reported mitogenome of L. boringii (KJ630505). Then, the positions and directions of protein coding genes (PCGs), ribosomal RNA genes (rRNAs), transfer RNA genes (tRNAs), and the control region (D-loop) were annotated using MITOS Web Server [31]. The sites and secondary structure of tRNAs were further inferred with the help of ARWEN and Scan-SE [32]. Visualization and circularization of the complete mitogenome were performed through the online server GeSeq [33]. Other analyses, such as nucleotide composition, AT and GC skew, and the determination of sequence genetic distances under Kimura’s two-parameter (K2P) model [34], were conducted through MEGA X [35]. Relative synonymous codon usage (RSCU), nucleotide diversity (Pi), and the ratio of the nonsynonymous substitution rate (Ka) and the synonymous substitution rate (Ks) were all calculated through DnaSP 6 [36].
2.3. Phylogenetic Analysis
The sequences used for phylogenetic reconstruction consisted of L. boringii (SZ population) and L. liui (WLY population) sequences we obtained in this study and another 27 mitogenomes that were downloaded from NCBI, including 2 mitogenomes of L. boringii (EM and PS populations), 1 mitogenome of L. liui (JLS population), and 24 mitogenomes of other 21 representative species within Megophryidae. Microhyla fissipes in the Microhylidae family was used as the outgroup. Each of the 13 PCGs was extracted from the dataset of 30 mitogenomes and manually checked, and all PCGs were then aligned using the inbuilt MUSCLE module in MEGA X and concatenated to make a combined PCG dataset. To uncover the population status and phylogenetic position of L. boringii and L. liui, the phylogenetic analyses were reconstructed using both the maximum likelihood (ML) method conducted in RAxML 8.0.2 [37] and the Bayesian inference (BI) method via MrBayes 3.2.7 [38]. Partitioning scheme and nucleotide substitution models for ML and BI phylogenetic analyses were selected using PartitionFinder 2 [39] based on the Akaike information criterion (AIC). The statistical confidence was assessed through a bootstrap test with 1000 replicates in ML trees and posterior probability calculation of the BI trees that under simultaneously run for 1.0 × 107 million generations, with sampling conducted every 1000 generations and discarding the initial 25% generations.
3. Results
3.1. Mitogenome Annotation and Nucleotide Composition
The assembled complete mitogenomes of L. boringii and L. liui were 17,100 and 17,501 bp in length, which were deposited in NCBI under accession numbers OP373724 and OP503540, respectively. The mitogenomes of both species had a typical gene organization, with 13 PCGs, 2 rRNAs, 23 tRNAs, and a D-loop region (Table 1; Figure 2). There was a total of 187 and 174 bp of intergenic nucleotides (IGNs) dispersed in 12 and 13 locations of two species, ranging from 1 to 108 bp and 1 to 99 bp in length, respectively (Table 1). Similar to other species in Megophryidae, the overall base composition of L. boringii was 31.5% T, 25.5% C, 15.3% G, and 27.7% A, while that of L. liui was 32.7% T, 24.3% C, 14.8% G, and 28.1% A (Table 2), indicating an A+T bias with greater A+T than G+C content (59.2% vs. 40.8% and 60.8% vs. 39.1%). Additionally, both the AT and GC skew were negative for the mitogenomes of all Megophryidae species, reflecting a general bias toward T and C base pairs (Table 2).

Table 1.
Characteristics of the mitogenome of L. boringii (LB) and L. liui (LL).
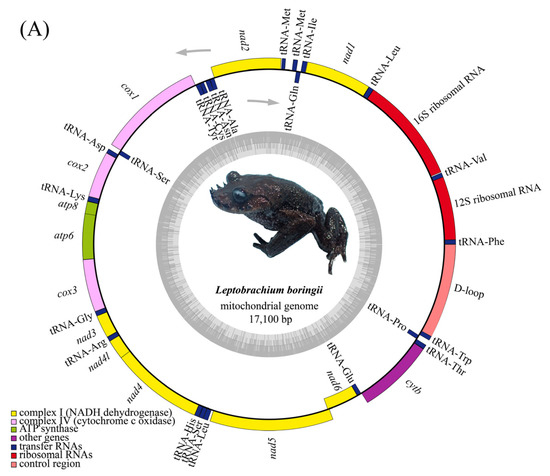
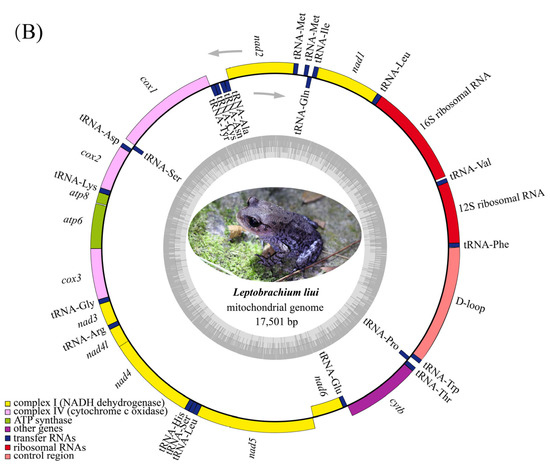
Figure 2.
Gene map of the mitogenome of L. boringii (A) and L. liui (B).
Table 2.
Base composition (in percentages) of the mitogenomes of three populations of L. boringii, two populations of L. liui, and another 21 species in Megophryidae.
3.2. Characteristics of rRNAs, tRNAs, and the Control Region
In both L. boringii and L. liui, there were 23 tRNAs interspersed in the whole mitogenome, ranging from 64 to 75 bp, with tRNACys being the shortest and tRNALeu the longest. Similarly to other congeners in the genus Leptobrachium, there was also a tandem duplication of tRNAMet that was separated by long IGNs (intergenic nucleotides, 108 bp in L. boringii and 99 bp in L. liui). There were two rRNAs with a total length of 2520 bp in L. boringii and 2500 bp in L. liui. The 16S rRNA was located between tRNAVal and tRNALeu with a length of 1580 bp in L. boringii and 1563 bp in L. liui, whereas the 12S rRNA was located between tRNAPhe and tRNAVal with 940 bp in L. boringii and 937 bp in L. liui. The non-coding domain of D-loop was located between tRNATrp and tRNAPhe, with a length of 1406 bp in L. boringii and 1815 bp in L. liui (Table 1). For both L. boringii and L. liui, most genes were encoded on the heavy (H) strand, except for the nad6 and eight tRNA genes (tRNAGln, tRNAAla, tRNAAsn, tRNACys, tRNATyr, tRNASer, tRNAGlu, and tRNAPro) encoded on the light (L) strand.
Among all the 13 PCGs, the shortest was the atp8 gene, being only 186 bp in L. boringii and 165 bp in L. liui, while the longest was nad5 at 1833 bp in L. boringii and 1830 bp in L. liui. Three PCGs (cox1, atp8, nad3) started with GTG, while the other ten PCGs used ATG as the initiation codon in L. boringii; however, in L. liui, by comparison, there was one more PCG (nad4) starting with GTG, and the remaining nine PCGs start with ATG. The termination codon usage in L. boringii and L. liui was diverse: complete codons were found to be used in seven PCGs (TAA for nad2, cox1, nad3, nad4l, and nad5; TGA for atp8; AGG for nad6) and incomplete codons by the six other PCGs (TA+ for nad1 and atp6; T++ for cox2, cox3, nd4, and cytb) in L. boringii, while in L. liui, a difference appeared in the usage of complete termination codons (TAA for nad2, cox1, nad3, and nad4l; CAT for atp8; AGG for nad5 and nad6). Nucleotide composition analysis of the 13 PCGs revealed that they shared similar patterns, except for the nad6 gene, which had unusual base proportions of T (35.1%), C (12.7%), G (33.7%), and A (18.4%) in L. boringii and T (35.5%), C (12.9%), G (32.9%), and A (18.6%) in L. liui, respectively; thus, the nad6 gene presents an extraordinary but positive GC skew (Table 3). There were several distinct but overlapping sites found in the mitogenomes of the two species: in L. boringii, these sites ranged from 1 to 28 bp and overlapped at 10 positions, occupying a total length of 50 bp, with the longest overlap (28 bp) located between the nad5 and nad6 genes; in L. liui, the overlapping sites were found dispersed in nine positions that range from 1 to 7 bp and a total length of 22 bp, with the longest (7 bp) lying between nad4l and nad4.
Table 3.
Nucleotide composition and skewness of different regions in the mitogenomes of L. boringii (LB) and L. liui (LL).
3.3. Codon Usage and Genetic Distances
The codon usage of the PCGs showed an extremely similar pattern that was shared between L. boringii and L. liui (Figure 3). The UCU (Ser1), CGA (Arg), UUA (Leu), and CAA (Gln) had the highest frequencies in both L. boringii and L. liui. RSCU analysis of both species indicated that six amino acids (Val, Pro, Thr, Ala, Arg, and Gly) were encoded by four synonymous codons, with the exceptions being Leu and Ser, encoded by six codons, and all others (Phe, Ile, Met, Tyr, His, Gln, Asn, Lys, Asp, Glu, Cys, and Trp) were encoded by two codons (Figure 3). Ka was generally less than Ks, and the average values between species pairs within 21 Megophryidae species ranged from 0 to 0.686 for Ka and from 0 to 6.2922 for Ks (Figure 4A). The Ka/Ks ratios of all the 13 PCGs were less than 1, with the highest Ka/Ks ratio (0.675) in atp8 (Figure 4B). None of the PCGs showed Ka/Ks ≥ 1, which indicates a generally negative or purifying selection. The atp8, atp6, and nad2 genes were found to have relatively fast evolutionary rates, whereas the cox1, cox3, and cytb genes have a relatively slow evolutionary rate.
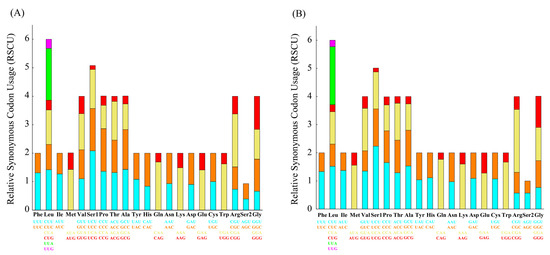
Figure 3.
Relative synonymous codon usage (RSCU) of mitogenome of L. boringii (A) and L. liui (B).
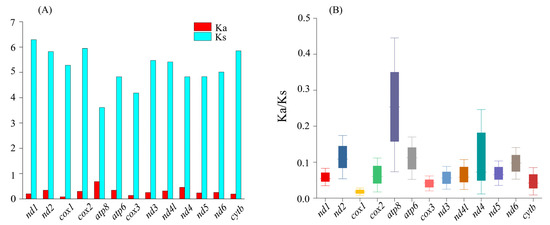
Figure 4.
(A) The ratio of synonymous (Ks) and non-synonymous (Ka) substitution, and (B) Ka/Ks of 13 PCGs among 21 species within Megophryidae.
The overall genetic distances of PCGs between two of the three populations of L. boringii were 4.82% (EM and PS), 4.22% (EM and SZ), and 4.19% (PS and SZ). While looking at each of the PCGs, atp8 had the highest genetic variance, followed by cox2, cox1, and nad2, and cox3 had the lowest genetic variance among the three populations (Figure 5A). As for L. liui, the overall genetic distance of PCGs between the two populations (JLS and WLY) was 7.09%, in which atp8 and nad4l had the highest and the lowest genetic variances, respectively (Figure 5A). In addition, when using the sliding window analysis along the concatenated PCG dataset, we found that the nucleotide diversity (Pi) was also highly variable among different gene sequences, ranging from 0.198 (cox1) to 0.352 (atp8) of 13 PCGs within the 21 species. Generally, the atp8, atp6, and nad2 genes had relatively high nucleotide diversity, while the cox1, cox2, and cox3 genes had comparatively low nucleotide diversity, no matter the examined sequences, which were grouped from (1) three populations of L. boringii and two populations of L. liui (Figure 5B), (2) four subgroups of Clade II (Figure 5C), or two major clades of Megophryidae (Figure 5D).
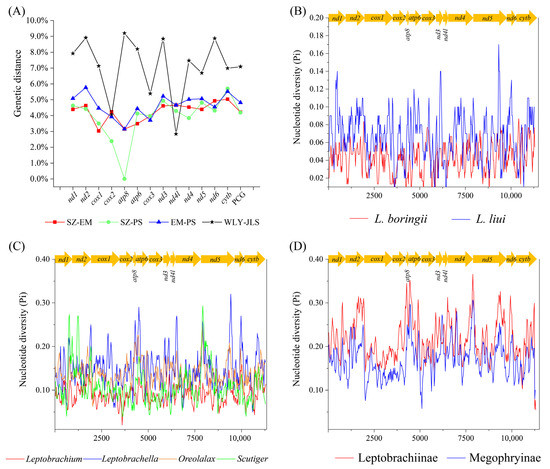
Figure 5.
(A) Genetic distances between different populations of L. boringii and L. liui, and nucleotide diversity (Pi) under sliding window analysis of (B) three populations of L. boringii and two populations of L. liui, (C) four groups of Clade II, and (D) two clades of Megophryidae.
3.4. Phylogenetic Analysis
The phylogenetic trees based on both ML and BI methods showed highly consistent topologies, with strong support values in most branches (Figure 6). The genus Leptobrachium was divided into two subgroups. L. boringii was recovered into the sister group of L. ailaonicum, and the three samples of L. boringii formed a monophyly, while the PS population branched first and the EM population and SZ population followed (obtained in this study). The two populations (WLY and JLS) of L. liui formed a monophylum and were recovered as the sister species of L. leishanese. According to the phylogenetic tree, the Megophryidae can be divided into two major clades corresponding to two subfamilies. Clade I (the subfamily Megophryinae) contains species of the following four genera: Atympanophrys, Megophrys, Brachytarsophrys, and Boulenophrys. Clade II (the subfamily Leptobrachiinae) can be further divided into four well-supported major groups in the following order: Leptobrachella (Group A) first, followed by Scutiger (Group B), then the sister groups Oreolalax (Group C) and Leptobrachium (Group D) (Figure 6).
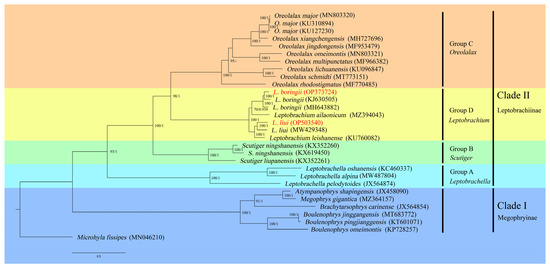
Figure 6.
Phylogenetic relationships within Megophryidae derived from ML method based on 13 PCGs. Note: The numbers on the branches represent the bootstrap values and posterior probabilities of ML/BI analyses. The number after species name is the GenBank accession number. Names in red show the phylogenetic positions of L. boringii and L. liui that we sequenced in this study.
4. Discussion
Mitochondrial DNA serves as an efficient molecular marker that has been widely applied in evolution-related studies for a variety of species [40]. Compared with relatively abundant and complex nuclear DNA, the mitochondrial genome has many favorable features, such as simple structure with conserved coding regions, low levels of recombination, multiple copy number, rapid evolutionary rate, and maternal inheritance [40,41]. Therefore, mitochondrial DNA has played a valuable role in reconstructing phylogenetic relationships, revealing population genetic structures, estimating divergence times, identifying relatedness between recently diverged species, etc. [29,42,43,44,45].
The traditional method for obtaining a mitogenome is the PCR-targeted method based on chain termination sequencing [46], which has greatly promoted the development of relevant studies. Although the PCR-targeted method is simple to operate, it is time-consuming and primer-dependent, and the mitogenomes combined based on different DNA fragments were usually uncomplete [27,28]. In recent years, the advent of next-generation sequencing (NGS) technology has revolutionized biological studies for producing thousands or even millions of DNA reads within a short period [47,48]. With the ever-increasing throughput and ever-decreasing costs with the development of NGS, more and more complete mitochondrial genomes have been reported and used as essential sources and optimal molecular markers for studies on evolution, phylogenetics, population genetics, and biological conservation [29,43,48,49,50,51]. Although previous studies [27,28] have already reported the mitogenomes of L. boringii based on the traditional PCR-targeted method, these mitogenomes were not as complete as what we assembled based on NGS in this study. Nevertheless, the gene organization, base composition pattern, and transcriptional direction were almost the same among the three mitogenomes of L. boringii, and they were also similar to those of other congeners [29,52], including L. liui that we sequenced and assembled in this study (Table 1, Table 2 and Table 3, Figure 2).
As a result of this study, we report and describe the characteristics of two newly sequenced mitogenomes of L. boringii and L. liui in more detail. For instance, codon usage bias is a phenomenon in which specific codons are used more frequently than other synonymous codons by certain organisms during the translation of genes to proteins. With rapid progress in whole-genome sequencing, analysis of codon usage bias at the genome level, rather than for a single gene or a set of genes, has been increasing [42]. The results of RSCU analyses indicate that in both L. boringii and L. liui, codons with A or T at the third position are always overused compared with other synonymous codons (Figure 3). The biased usage of AT nucleotides is also reflected in the form of codon usage, where the highly frequently used codons, UCU (Ser1), CGA (Arg), UUA (Leu), and CAA (Gln), all end with A or T, which might also partly contribute to the higher A+T content.
The positive selection analyses based on the PCGs of mitogenome can shed light on the question of whether natural selection affected the functional characteristics of the mitochondrion [53]. The ratio of Ka and Ks is a popular proxy to detect adaptive evolution from viruses to humans [54]. It is considered that Ka/Ks > 1 indicates positive selection, Ka/Ks = 1 neutrality, and Ka/Ks < 1 negative or purifying selection [55]. The Ka/Ks value from each of the PCGs within Megophryidae was less than 1 (Figure 4), which indicates that the overall evolution pattern of the mitogenomes of Megophryidae tend to be conservative in maintaining the functions of regularly generated proteins [56]. Interestingly, the Ka/Ks of each of the PCGs indicates that the evolutionary rates are relatively fast for atp8, atp6, and nad2 and relatively slow for cox1, cox3, and cytb (Figure 4). This pattern was also revealed in the nucleotide diversity analyses based on different PCGs or from different examined groups under sliding windows (Figure 5).
The previously known mitogenomes of L. boringii and L. liui from other populations [27,28,29], and two newly sequenced mitogenomes in this study, together, provided us with an excellent opportunity to examine the intraspecific and interspecific genetic distances among the species of Leptobrachium. The intraspecific genetic distances of L. boringii were generally greater than 4%, which means the three examined populations are highly differentiated. This trend was also revealed in other studies. For instance, Yang et al. [57] found that L. boringii in the Sichuan and Hunan Provinces had significant genetic differentiation with relatively poor gene flow, and the genetic distance between them was 4.7% based on 1097 bp of cytb genes. Interestingly, our study further showed that the differentiation pattern of L. boringii is not straightforward along the current geographic distances. The PS population (in Chongqing Province) was between the EM (in Sichuan Province) and SZ population (in Hunan Province) from the geographic point of view; however, it divided first and allowed the remaining far-distanced EM and SZ populations to group together in the phylogenetic tree (Figure 6). This suggests the phylogeographic pattern of L. boringii among its ranges is shaped by multiple factors. Comparatively, the intraspecific genetic distance between the two populations of L. liui was even greater (7.09%) and almost reached that of the interspecific genetic distance between L. liui and L. leishanensis (7.85%). Although we could identify that the sample we sequenced was a subspecies (L. liui yaoshanensis) due to its four keratinized spines [22], we were unable to identify whether or not the JLS population was from another subspecies (L. liui liui) because no morphological traits were mentioned in the literature [29] and, additionally, whether the JLS population was in the boundary area of two subspecies. Even so, our results show that the intraspecific genetic distance of L. liui is substantially high; thus, further taxonomic studies are of worth. The interspecific genetic distance between L. boringii and L. liui was relatively high, as they were found in two different subgroups of Leptobrachium (Figure 5), which is consistent with another study based on 2 mitochondrial DNA markers (cytb and nad4) and 11 microsatellite loci [58].
While using 27 mitogenome sequences available from NCBI and 2 newly obtained in this study as the ingroups, with Microhyla fissipes in the Microhylidae family as the outgroup, a phylogenetic tree within Megophryidae based on mitogenomes was able to be reconstructed in this study (Figure 5). The family Megophryidae was divided into two clades (Clade I and II) corresponding to two subfamilies, Megophryinae and Leptobrachiinae, respectively. Although the species involved here are still limited, the division of two subfamilies is supported. Leptobrachiinae was divided into four groups corresponding to four genera and well supported based on the values: the genus Leptobrachella divided first and then Scutiger, followed by the sister group of Leptobrachium and Oreolalax. However, this is not consistent with a previous study based on three mitochondrial and nine nuclear genes, which showed that Leptobrachella divided first, followed by Leptobrachium, leaving Scutiger and Oreolalax to form a sister group [5]. This is a reminder that the intergeneric phylogeny within Leptobrachiinae requires further study for verification. When it comes to the phylogenetic relationships within Leptobrachium, our study shows that two lineages can be distinguished: one with L. boringii and L. ailaonicum, and the other one formed by L. liui and L. leishanense. The relationship of the above two lineages is consistent with the traditional distinction of two species groups according to morphological characteristics, especially the number of keratinized spines (10–48 in L. boringii species group vs. 2–6 in L. liui species group, [22]). However, this relationship is not well supported by previous studies using relatively limited mitochondrial DNA sequences. For instance, Rao et al. [8] and Matsui et al. [14] revealed a (L. ailaonicum, (L. boringii, (L. liui, L. leishanense))) relationship based on 16S rRNA, nad4, and cytb (a total of 2566 bp), and 12S rRNA, tRNAVal, and 16S rRNA (a total of 2009 bp), respectively. Chen et al. [59] recovered a (L. boringii, (L. ailaonicum, (L. liui, L. leishanense))) relationship based on 1914 bp of 12S rRNA, tRNAVal, and 16S rRNA sequences. Although the phylogenetic relationships would change along with the differences in the DNA markers used, it has been generally recognized that the use of a complete mitogenome is superior to individual genes in uncovering evolutionary relationships [60,61]. However, it has to be noted that the bootstrap value of the node of L. ailaonicum and L. boringii in our study was still not so supportive (only 70), which could also be treated and collapsed as an unresolved polytomy with a (L. ailaonicum, L. boringii (L. liui, L. leishanense)) relationship. In a word, the whole picture of the evolutionary history within the groups of Megophryidae is still as presented until more and more mitogenome data, such as for the two species reported in this study, become available in the future.
Author Contributions
Conceptualization, H.-M.X. and W.-S.J.; methodology, Q.Z. and W.-S.J.; software, Q.Z. and M.-Y.Z.; validation, W.-S.J. and M.-Y.Z.; formal analysis, Q.Z. and W.-S.J.; investigation, X.-Y.L. and J.-X.W.; resources, Z.-R.G. and Y.L.; data curation, Q.Z.; writing—original draft preparation, Q.Z.; writing—review and editing, H.-M.X. and W.-S.J.; visualization, Q.Z.; supervision, W.-S.J.; project administration, W.-S.J.; funding acquisition, W.-S.J. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Talent Project of Hunan Provincial Science and Technology Department (2020RC3057), National Natural Science Foundation of China (32060128), Zhilan foundation (2020040371B), and Opening Projects of “Graduate Student” of Jishou University (JGY2022100, DNGC2212).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The assembled mitogenome sequences were deposited in NCBI (https://www.ncbi.nlm.nih.gov/ (accessed on 1 October 2022)) with accession numbers: OP373724 and OP503540. All data generated by this study are available from the corresponding author upon reasonable request.
Acknowledgments
We thank Bi-Wu Qin (National Nature Reserve of Badagongshan) and Gui-Xiang Xie (Zhangjiajie National Forest Park) for their help and support during field work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mahony, S.; Kamei, R.G.; Teeling, E.C.; Biju, S.D. Cryptic diversity within the Megophrys major species group (Amphibia: Megophryidae) of the Asian Horned Frogs: Phylogenetic perspectives and a taxonomic revision of South Asian taxa, with descriptions of four new species. Zootaxa 2018, 4523, 1–96. [Google Scholar] [CrossRef] [PubMed]
- Cannatella, D.C. A Phylogeny of Primitive Frogs (Archaeobatrachians) (Cladistics, Taxonomy, Morphology, Systematics, Evolution). Ph.D. Thesis, University of Kansas, Lawrence, Kansas, 1985. [Google Scholar]
- Ford, L.S.; Cannatella, D.C. The Major Clades of Frogs. Herpetol. Monogr. 1993, 7, 94–117. [Google Scholar] [CrossRef]
- Frost, D.R.; Grant, T.; Faivovich, J.; Bain, R.H.; Haas, A.; Haddad, C.F.B.; Desa, R.O.; Channing, A.; Wilkinson, M.; Donnellan, S.C.; et al. The amphibian tree of life. Bull. Am. Mus. Nat. Hist. 2006, 297, 1–370. [Google Scholar] [CrossRef]
- Pyron, R.A.; Wiens, J.J. A large-scale phylogeny of Amphibia including over 2800 species, and a revised classification of extant frogs, salamanders, and caecilians. Mol. Phylogenet. Evol. 2011, 61, 543–583. [Google Scholar] [CrossRef]
- AmphibiaWeb. Available online: https://amphibiaweb.org/ (accessed on 8 February 2023).
- Fei, L.; Ye, C.Y.; Jiang, J.P.; Huang, Y.Z. Atlas of Amphibians in China (Field Edition), 1st ed.; Henan Science and Technology Press: Zhengzhou, China, 2020; pp. 254–262. [Google Scholar]
- Rao, D.Q.; Wilkinson, J.A. Phylogenetic relationships of the mustache toads inferred from mt DNA sequences. Mol. Phylogenet. Evol. 2008, 46, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Fei, L.; Ye, C.Y.; Jiang, J.P. Colored Atlas of Chinese Amphibians, 1st ed.; Sichuan Publishing House of Science and Technology: Chengdu, China, 2010; pp. 162–167. [Google Scholar]
- Tian, W.; Hu, S. Taxonomical studies on the primitive anurans of the Hengduan Mountains, with descriptions of a new subfamily and subdivision of Bombina. Acta Herpetol. Sinica 1985, 4, 219–224. [Google Scholar]
- Dubois, A.; Ohler, A. A new species of Leptobrachium (Vibrissaphora) from northern Vietnam, with a review of the taxonomy of the genus Leptobrachium (Pelobatidae, Megophryinae). Dumerilia 1998, 4, 1–32. [Google Scholar]
- Zhao, E.M.; Adler, K. Herpetology of China, 1st ed.; Society for the Study of Amphibians and Reptiles: Oxford, MI, USA, 1993; pp. 1–522. [Google Scholar]
- Zheng, Y.C.; Li, S.; Fu, J. A phylogenetic analysis of the frog genera Vibrissaphora and Leptobrachium, and the correlated evolution of nuptial spine and reversed sexual size dimorphism. Mol. Phylogenet. Evol. 2008, 46, 695–707. [Google Scholar] [CrossRef]
- Matsui, M.; Hamidy, A.; Murphy, R.W.; Khonsue, W.; Yambun, P.; Shimada, T.; Ahmad, N.; Belabut, D.M.; Jiang, J.P. Phylogenetic relationships of megophryid frogs of the genus Leptobrachium(Amphibia, Anura) as revealed by mtDNA gene sequences. Mol. Phylogenet. Evol. 2010, 56, 259–272. [Google Scholar] [CrossRef]
- Gao, J.M.; Zhang, Y.D. Retinal organization in Vibrissaphora liui. Acta Zool. Sin. 1996, 42, 405–412. [Google Scholar]
- Zheng, Y.C.; Zeng, X.M. A review of study on moustache toads, Vibrissaphora (Anura: Megophryidae). Sichuan J. Zool. 2003, 22, 268–271. [Google Scholar]
- Zhang, W.Y.; Lu, Y.Y.; Shi, J.S.; Zhu, L.; Li, P.P. Microstructure and Sub-Microstructure of the Keratinized Nuptial Spines of Male Leptobrachium boringii (Liu, 1945). Sichuan J. Zool. 2019, 38, 186–193. [Google Scholar]
- Hudson, C.M.; He, X.J.; Fu, J.Z. Keratinized Nuptial Spines Are Used for Male Combat in the Emei Moustache Toad (Leptobrachium boringii). Asian Herpetol. Res. 2011, 2, 142–148. [Google Scholar]
- Zheng, Y.C.; Deng, D.C.; Li, S.Q.; Fu, J.Z. Aspects of the breeding biology of the Omei mustache toad (Leptobrachium boringii): Polygamy and paternal care. Amphib. -Reptil. 2010, 31, 183–194. [Google Scholar] [CrossRef]
- Hudson, C.M.; Fu, J. Male-biased sexual size dimorphism, resource defense polygyny, and multiple paternity in the Emei moustache toad (Leptobrachium boringii). PLoS ONE 2013, 8, e67502. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Ren, Y.D.; Zhang, D.R.; Jiang, H.; Wang, Z.K.; Li, X.Y.; Rao, D.Q. Chromosome-level assembly of the mustache toad genome using third-generation DNA sequencing and Hi-C analysis. GigaScience 2019, 8, giz114. [Google Scholar] [CrossRef] [PubMed]
- Fei, L.; Hu, S.Q.; Ye, C.Y.; Huang, Y.Z. Amphibia. In Fauna Sinica, 1st ed.; Fauna Sinica Editorial Board; Science Press: Beijing, China, 2009; Volume 2, pp. 81–485. [Google Scholar]
- Liu, B.H. Vibrissaphora liui in Jiulongshan Mountain, Zhejiang province. Chin. J. Wildl. 1987, 6, 22–23. [Google Scholar]
- IUCN SSC Amphibian Specialist Group. Leptobrachium boringii. In The IUCN Red List of Threatened Species 2020. 2020. [Google Scholar] [CrossRef]
- Gao, Z.W.; Qian, T.Y.; Jiang, J.P.; Hou, D.J.; Deng, X.J.; Yang, D.D. Species diversity and distribution of amphibians and reptiles in Hunan Province, China. Biodivers. Sci. 2022, 30, 101–115. [Google Scholar] [CrossRef]
- IUCN SSC Amphibian Specialist Group. Leptobrachium liui. In The IUCN Red List of Threatened Species 2020. 2020. [Google Scholar] [CrossRef]
- Xu, Q.P.; Liu, S.L.; Wan, R.Z.; Yue, B.S.; Zhang, X.Y. The complete mitochondrial genome of the Vibrissaphora boringii (Anura: Megophryidae). Mitochondrial DNA Part A 2016, 27, 758–759. [Google Scholar] [CrossRef]
- Li, Y.W.; Li, C.; Chen, Q.L.; Liu, Z.H.; Shen, Y.J. The mitochondrial genome for tadpole of Emei moustache toad (Leptobrachium boringii: Anura:Megophryidae) from the Southwest China and its phylogenetic analysis. Mitochondrial DNA Part B Resour. 2019, 4, 265–266. [Google Scholar] [CrossRef]
- Xiang, Z.Y.; Zhang, C.Y.; Zheng, W.C.; Yu, S.S.; Zhang, L.; Ding, G.H. Characterization of the complete mitochondrial genome of the Chong’an Moustache Toad, Leptobrachium liui (Pope, 1947) with a phylogenetic analysis of Megophryidae. Mitochondrial DNA Part B Resour. 2021, 6, 1061–1063. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, D.; Patrick, M.; Guillaume, S. NOVOPlasty: De Novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Zardoya, R.; Meyer, A. Phylogenetic performance of mitochondrial protein-coding genes in resolving relationships among vertebrates. Mol. Biol. Evol. 1996, 13, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.Y.; Chang, H.H.; Yuan, H.; Huang, Y.; Lu, H.M.; Li, X.; Gou, X.C. Comparative mitochondrial genomes of four species of Sinopodisma and phylogenetic implications (Orthoptera, Melanoplinae). ZooKeys 2020, 969, 23–42. [Google Scholar]
- Bao, X.L.; Cui, L.; Li, D.Y.; Fan, X.L.; Yang, M.Y.; Yang, D.Y.; Ni, Q.Y.; Yao, Y.F.; Xu, H.L.; Zeng, B.; et al. The near complete mitochondrial genome of Oreolalax schmidti (Anura: Megophryidae). Mitochondrial DNA Part B Resour. 2020, 5, 3536–3537. [Google Scholar] [CrossRef]
- Shu, G.C.; Yu, M.; He, Z.P.; Xie, F.; Liang, X.X. Complete mitochondrial genome of the Alpine Metacarpal-tubercled Toad Leptobrachella alpina (Amphibia, Anura, Megophryidae). Mitochondrial DNA B Resour. 2021, 6, 3242–3243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.F.; Miao, G.P.; Hu, S.J.; Sun, Q.; Ding, H.W.; Ji, Z.C.; Guo, P.; Yan, S.B.; Wang, C.R.; Kan, X.Z.; et al. Quantification and evolution of mitochondrial genome rearrangement in Amphibians. BMC Ecol. Evol. 2021, 21, 19. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef]
- Ye, F.; Samuels, D.C.; Clark, T.; Guo, Y. High-throughput sequencing in mitochondrial DNA research. Mitochondrion 2014, 17, 157–163. [Google Scholar] [CrossRef]
- Jex, A.R.; Hall, R.S.; Littlewood, D.T.J.; Gasser, R.B. An integrated pipeline for next-generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res. 2010, 38, 522–533. [Google Scholar] [CrossRef]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Lan, X.Y.; Luo, Q.H.; Gu, Z.R.; Zhou, Q.; Zhang, M.Y.; Zhang, Y.X.; Jiang, W.S. Characterization, Comparison of Two New Mitogenomes of Crocodile Newts Tylototriton (Caudata: Salamandridae), and Phylogenetic Implications. Genes 2022, 13, 1878. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.X.; Shu, G.C.; Wang, B.; Jiang, J.P.; Cheng, L.; Xie, F. Complete mitochondrial genome of the Leishan moustache toad, Vibrissaphora leishanensis (Anura: Megophryidae). Mitochondrial DNA B Resour. 2016, 1, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D.; Labthavikul, S.T.; Otey, C.R.; Arnold, F.H. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. USA 2006, 103, 5869–5874. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Shiu, S.H.; Li, W.H. The nonsynonymous/synonymous substitution rate ratio versus the radical/conservative replacement rate ratio in the evolution of mammalian genes. Mol. Biol. Evol. 2007, 24, 2235–2241. [Google Scholar] [CrossRef]
- Yang, Y.W. The Genetic Diversity and Geographical Differentiation of V. boringii. Master’s Thesis, Guizhou Normal University, Guizhou, China, 2014. [Google Scholar]
- Hu, M.L. Research on Population Demographic History of Four Species in Vibrissaphora in Mountain Region of South China. Master’s Thesis, Central China Normal University, Wuhan, China, 2015. [Google Scholar]
- Chen, W.C.; Zhang, W.; Zhou, S.C.; Li, N.; Huang, Y.; Mo, Y.M. Insight into the validity of Leptobrachium guangxiense (Anura: Megophryidae): Evidence from mitochondrial DNA sequences and morphological characters. Zootaxa 2013, 3641, 31–40. [Google Scholar] [CrossRef]
- Hou, X.J.; Lin, H.D.; Tang, W.Q.; Liu, D.; Han, C.C.; Yang, J.Q. Complete mitochondrial genome of the freshwater fish Acrossocheilus longipinnis (Teleostei: Cyprinidae): Genome characterization and phylogenetic analysis. Biologia 2020, 75, 1871–1880. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Zhou, Q.; Xiang, H.M.; Wang, J.X.; Lan, X.Y.; Luo, Q.H.; Jiang, W.S. Complete mitochondrial genome of Rectoris luxiensis (Teleostei, Cyprinidae): Characterisation and phylogenetic implications. Biodivers. Data J. 2023, 11, e96066. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).