
Network-Based and Machine-Learning Approaches Identify Diagnostic and Prognostic Models for EMT-Type Gastric Tumors
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Data Analysis and Visualization
2.3. Evaluation of ACRG Subtypes
2.4. Weighted Gene Co-Expression Network Analysis and Motif Identification
2.5. Assessment of Diagnostic and Prognostic Values of the RNAs
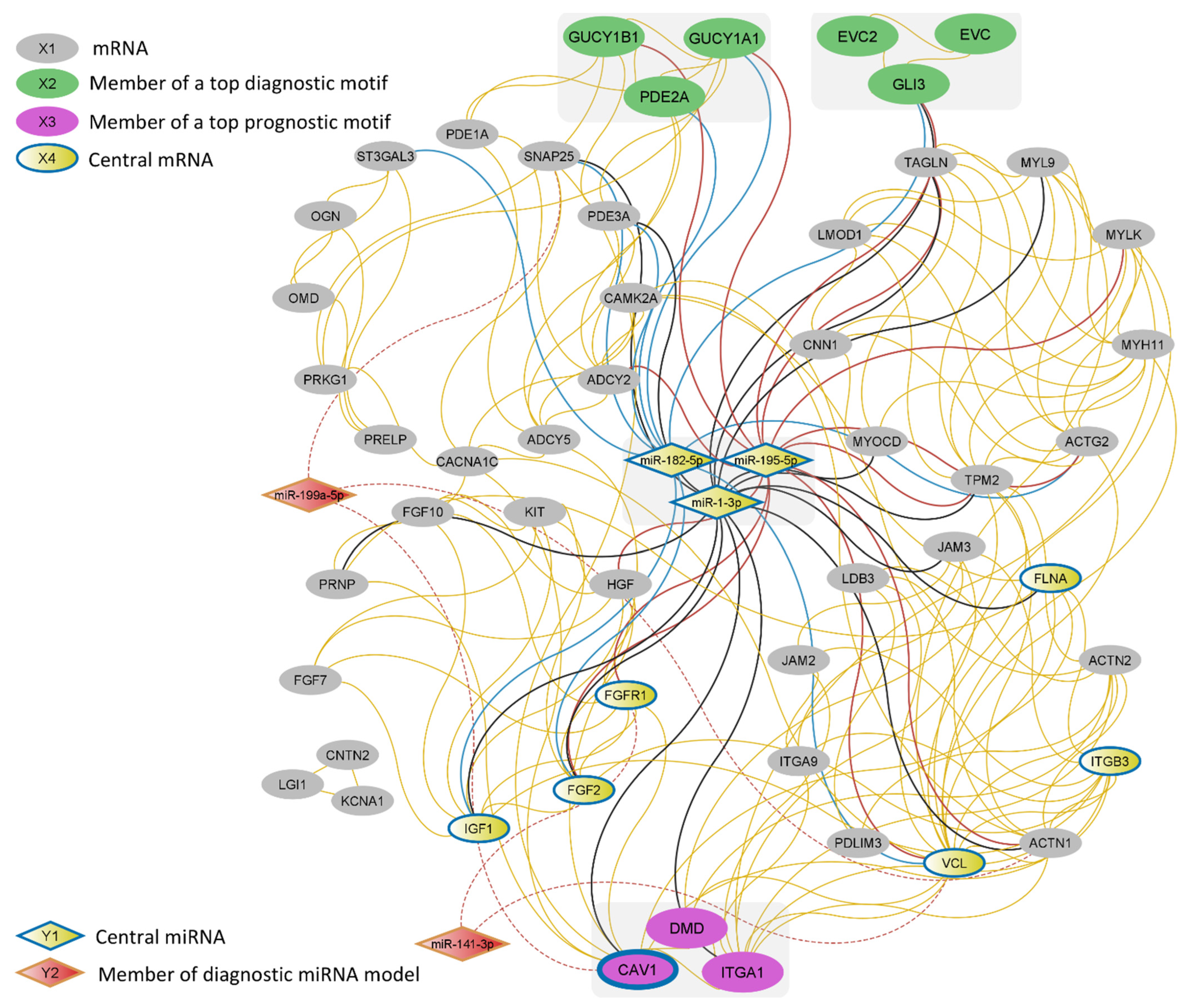
2.6. MiRNA-mRNA Network Construction
3. Results
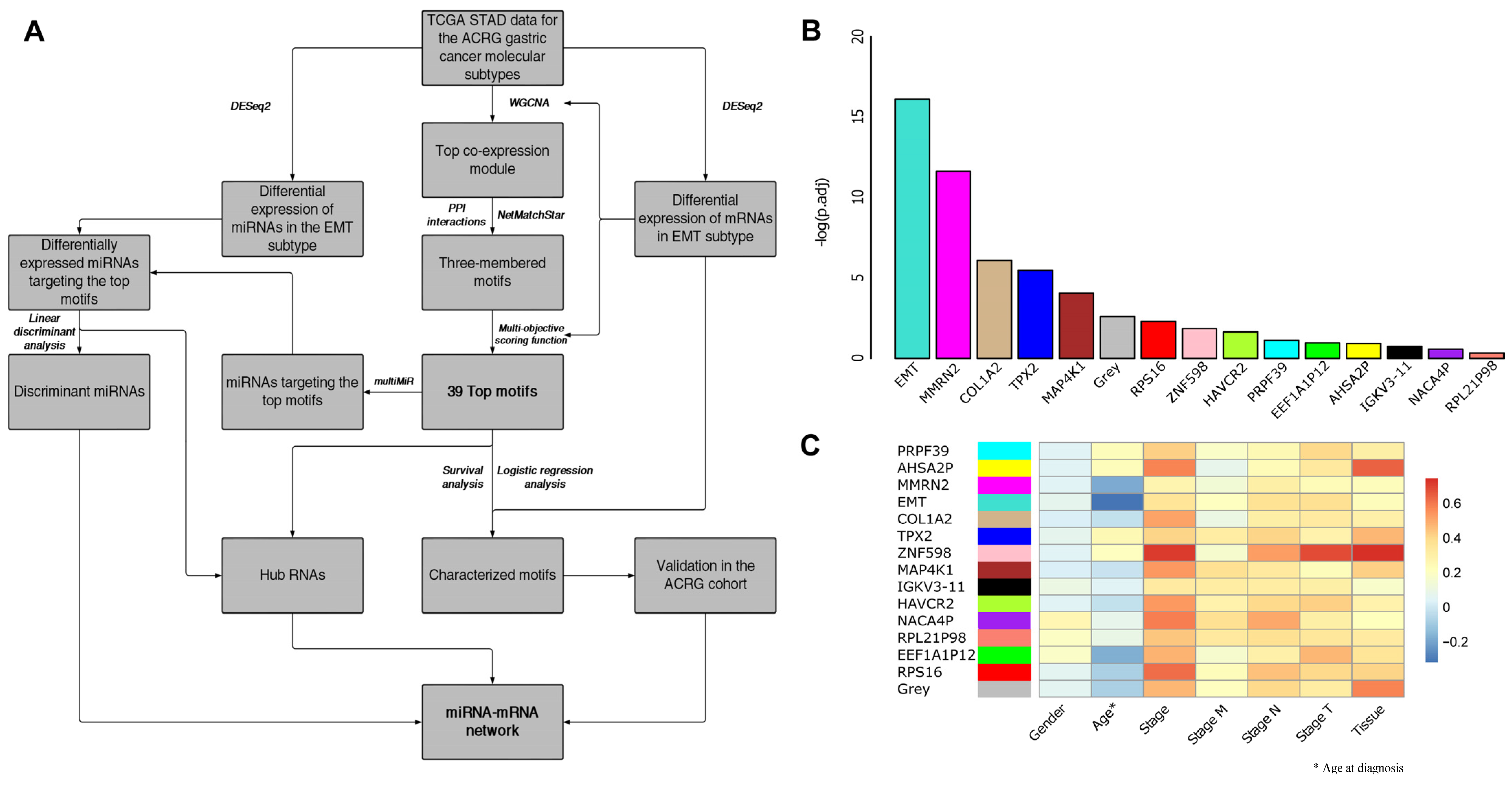
3.1. EMT-Type Gastric Cancer Displays a Distinct Transcriptional Profile
3.2. WGCNA and Motif Ranking Identify 39 Core mRNA Motifs
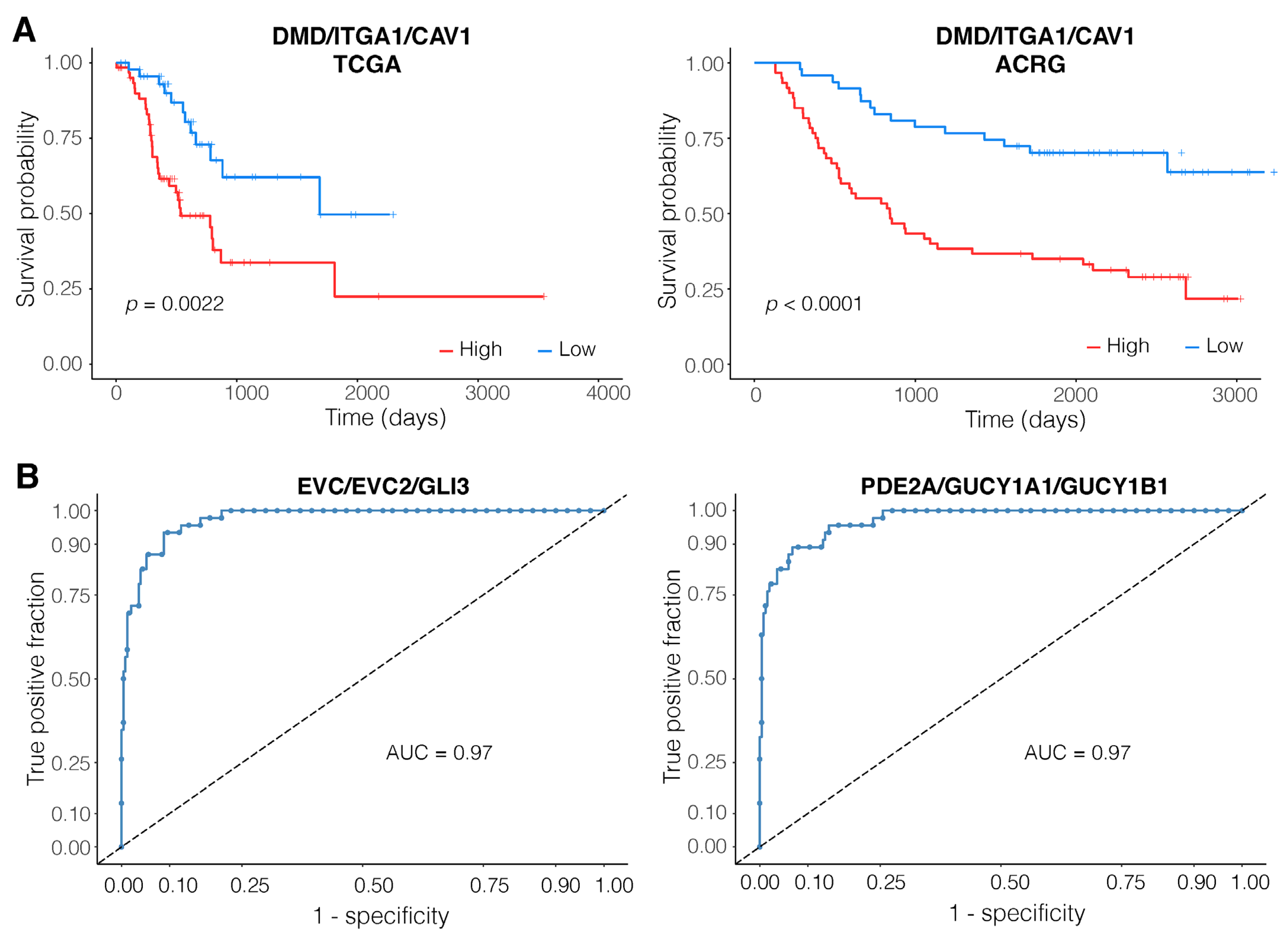
3.3. Expression of the DMD/ITGA1/CAV1 Motif Is a Strong Predictor of Patient Survival
3.4. EVC/EVC2/GLI3 and PDE2A/GUCY1A1/GUCY1B1 Are Robust Diagnostic Motifs
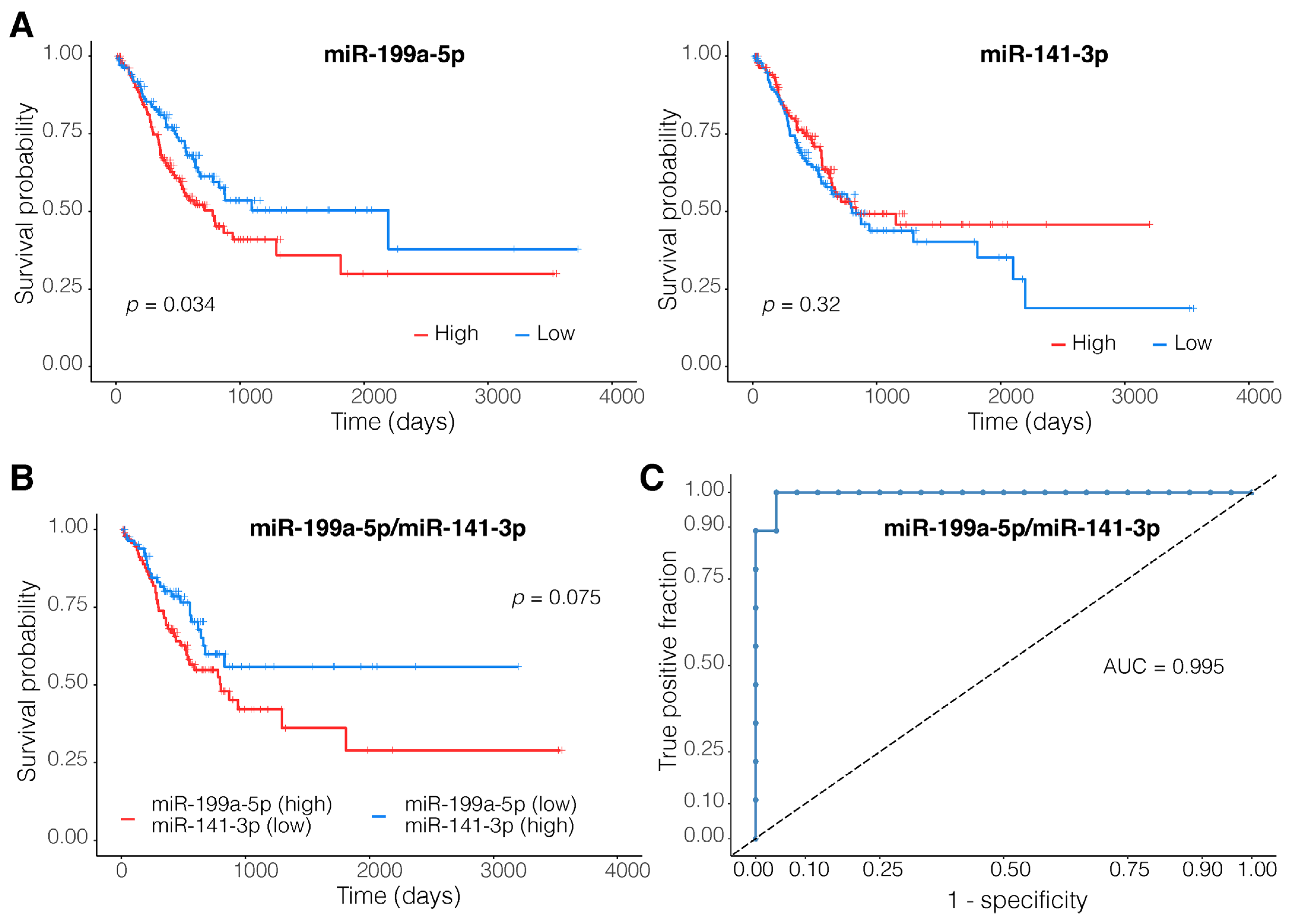
3.5. A Two-Membered miRNA Model Accurately Distinguishes EMT-Type Tumors from Other Gastric Tumors
4. Discussion
4.1. Poor Outcomes for Patients with High Expressions of DMD/ITGA1/CAV1 Motif
4.2. The EVC/EVC2/GLI3 Motif Performs Well Both as a Diagnostic and a Prognostic Marker
4.3. PDE2A/GUCY1A1/GUCY1B1—A Strong Diagnostic Marker
4.4. MiR-199a-5p and miR-141-3p Dysregulations Are Associated with Tumor Invasiveness
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Ho, S.W.T.; Tan, P. Dissection of Gastric Cancer Heterogeneity for Precision Oncology. Cancer Sci. 2019, 110, 3405–3414. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, J.; Huang, W.; Weng, S.; Wang, B.; Chen, Y.; Wang, H. Development and Validation of a Hypoxia-Immune-Based Microenvironment Gene Signature for Risk Stratification in Gastric Cancer. J. Transl. Med. 2020, 18, 201. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Sanjeevaiah, A.; Cheedella, N.; Hester, C.; Porembka, M.R. Gastric Cancer: Recent Molecular Classification Advances, Racial Disparity, and Management Implications. J. Oncol. Pract. 2018, 14, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Laurén, P. The Two Histological Main Types of Gastric Carcinoma: Diffuse and so-called Intestinal-Type Carcinoma. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- Serra, O.; Galán, M.; Ginesta, M.M.; Calvo, M.; Sala, N.; Salazar, R. Comparison and Applicability of Molecular Classifications for Gastric Cancer. Cancer Treat. Rev. 2019, 77, 29–34. [Google Scholar] [CrossRef]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.-M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular Analysis of Gastric Cancer Identifies Subtypes Associated with Distinct Clinical Outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Lee, J.; Cristescu, R.; Kim, K.-M.; Kim, K.; Kim, S.T.; Park, S.H.; Kang, W.K. Development of Mesenchymal Subtype Gene Signature for Clinical Application in Gastric Cancer. Oncotarget 2017, 8, 66305–66315. [Google Scholar] [CrossRef]
- Ooki, A.; Yamaguchi, K. The Dawn of Precision Medicine in Diffuse-Type Gastric Cancer. Ther. Adv. Med. Oncol. 2022, 14, 175883592210830. [Google Scholar] [CrossRef]
- Zhang, Z.; Hernandez, K.; Savage, J.; Li, S.; Miller, D.; Agrawal, S.; Ortuno, F.; Staudt, L.M.; Heath, A.; Grossman, R.L. Uniform Genomic Data Analysis in the NCI Genomic Data Commons. Nat. Commun. 2021, 12, 1226. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Ideker, T.; Ozier, O.; Schwikowski, B.; Siegel, A.F. Discovering Regulatory and Signalling Circuits in Molecular Interaction Networks. Bioinformatics 2002, 18, S233–S240. [Google Scholar] [CrossRef]
- Tang, J.; Kong, D.; Cui, Q.; Wang, K.; Zhang, D.; Gong, Y.; Wu, G. Prognostic Genes of Breast Cancer Identified by Gene Co-Expression Network Analysis. Front. Oncol. 2018, 8, 374. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Karimi, M.R.; Karimi, A.H.; Abolmaali, S.; Sadeghi, M.; Schmitz, U. Prospects and Challenges of Cancer Systems Medicine: From Genes to Disease Networks. Brief. Bioinform. 2022, 23, bbab343. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Rinnone, F.; Micale, G.; Bonnici, V.; Bader, G.D.; Shasha, D.; Ferro, A.; Pulvirenti, A.; Giugno, R. NetMatchStar: An Enhanced Cytoscape Network Querying App. F1000Research 2015, 4, 479. [Google Scholar] [CrossRef]
- Alon, U. Network Motifs: Theory and Experimental Approaches. Nat. Rev. Genet. 2007, 8, 450–461. [Google Scholar] [CrossRef]
- Khan, F.M.; Marquardt, S.; Gupta, S.K.; Knoll, S.; Schmitz, U.; Spitschak, A.; Engelmann, D.; Vera, J.; Wolkenhauer, O.; Pützer, B.M. Unraveling a Tumor Type-Specific Regulatory Core Underlying E2F1-Mediated Epithelial-Mesenchymal Transition to Predict Receptor Protein Signatures. Nat. Commun. 2017, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Ordway, B.; Rafiei, I.; Borad, P.; Fang, B.; Koomen, J.L.; Zhang, C.; Yoder, S.; Johnson, J.; Damaghi, M. Integrative Analysis of Breast Cancer Cells Reveals an Epithelial-Mesenchymal Transition Role in Adaptation to Acidic Microenvironment. Front. Oncol. 2020, 10, 304. [Google Scholar] [CrossRef] [PubMed]
- Anaya, J. OncoLnc: Linking TCGA Survival Data to MRNAs, MiRNAs, and LncRNAs. PeerJ Comput. Sci. 2016, 2, e67. [Google Scholar] [CrossRef]
- Thompson, J.A.; Tan, J.; Greene, C.S. Cross-Platform Normalization of Microarray and RNA-Seq Data for Machine Learning Applications. PeerJ 2016, 4, e1621. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, A.; Matsuzaki, J.; Yamamoto, Y.; Yoneoka, Y.; Takahashi, K.; Shimizu, H.; Uehara, T.; Ishikawa, M.; Ikeda, S.; Sonoda, T.; et al. Integrated Extracellular MicroRNA Profiling for Ovarian Cancer Screening. Nat. Commun. 2018, 9, 4319. [Google Scholar] [CrossRef] [PubMed]
- Ru, Y.; Kechris, K.J.; Tabakoff, B.; Hoffman, P.; Radcliffe, R.A.; Bowler, R.; Mahaffey, S.; Rossi, S.; Calin, G.A.; Bemis, L.; et al. The MultiMiR R Package and Database: Integration of MicroRNA–Target Interactions along with Their Disease and Drug Associations. Nucleic Acids Res. 2014, 42, e133. [Google Scholar] [CrossRef] [PubMed]
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Jalili, M.; Salehzadeh-Yazdi, A.; Asgari, Y.; Arab, S.S.; Yaghmaie, M.; Ghavamzadeh, A.; Alimoghaddam, K. CentiServer: A Comprehensive Resource, Web-Based Application and R Package for Centrality Analysis. PLoS ONE 2015, 10, e0143111. [Google Scholar] [CrossRef] [PubMed]
- Ashtiani, M.; Salehzadeh-Yazdi, A.; Razaghi-Moghadam, Z.; Hennig, H.; Wolkenhauer, O.; Mirzaie, M.; Jafari, M. A Systematic Survey of Centrality Measures for Protein-Protein Interaction Networks. BMC Syst. Biol. 2018, 12, 80. [Google Scholar] [CrossRef]
- Yeger-Lotem, E.; Sattath, S.; Kashtan, N.; Itzkovitz, S.; Milo, R.; Pinter, R.Y.; Alon, U.; Margalit, H. Network Motifs in Integrated Cellular Networks of Transcription–Regulation and Protein–Protein Interaction. Proc. Natl. Acad. Sci. USA 2004, 101, 5934–5939. [Google Scholar] [CrossRef]
- Latora, V.; Marchiori, M. Efficient Behavior of Small-World Networks. Phys. Rev. Lett. 2001, 87, 198701. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Ye, X.; Simon, S. The Integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Gharibi, A.; La Kim, S.; Molnar, J.; Brambilla, D.; Adamian, Y.; Hoover, M.; Hong, J.; Lin, J.; Wolfenden, L.; Kelber, J.A. ITGA1 Is a Pre-Malignant Biomarker That Promotes Therapy Resistance and Metastatic Potential in Pancreatic Cancer. Sci. Rep. 2017, 7, 10060. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Myint, P.K.; Ito, A.; Appiah, M.G.; Darkwah, S.; Kawamoto, E.; Shimaoka, M. Integrin-Ligand Interactions in Inflammation, Cancer, and Metabolic Disease: Insights Into the Multifaceted Roles of an Emerging Ligand Irisin. Front. Cell Dev. Biol. 2020, 8, 588066. [Google Scholar] [CrossRef] [PubMed]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Zhou, R.; Zhao, Y.; Wu, G. Integrin A6/Akt/Erk Signaling Is Essential for Human Breast Cancer Resistance to Radiotherapy. Sci. Rep. 2016, 6, 33376. [Google Scholar] [CrossRef]
- Zang, D.; Zhang, C.; Li, C.; Fan, Y.; Li, Z.; Hou, K.; Che, X.; Liu, Y.; Qu, X. LPPR4 Promotes Peritoneal Metastasis via Sp1/Integrin α/FAK Signaling in Gastric Cancer. Am. J. Cancer Res. 2020, 10, 1026–1044. [Google Scholar]
- Yan, H.; Zheng, C.; Li, Z.; Bao, B.; Yang, B.; Hou, K.; Qu, X.; Xiao, J.; Che, X.; Liu, Y. NPTX1 Promotes Metastasis via Integrin/FAK Signaling in Gastric Cancer. Cancer Manag. Res. 2019, 11, 3237–3251. [Google Scholar] [CrossRef]
- Wary, K.K.; Mariotti, A.; Zurzolo, C.; Giancotti, F.G. A Requirement for Caveolin-1 and Associated Kinase Fyn in Integrin Signaling and Anchorage-Dependent Cell Growth. Cell 1998, 94, 625–634. [Google Scholar] [CrossRef]
- Wang, X.; Lu, B.; Dai, C.; Fu, Y.; Hao, K.; Zhao, B.; Chen, Z.; Fu, L. Caveolin-1 Promotes Chemoresistance of Gastric Cancer Cells to Cisplatin by Activating WNT/β-Catenin Pathway. Front. Oncol. 2020, 10, 46. [Google Scholar] [CrossRef]
- Nam, K.H.; Lee, B.L.; Park, J.H.; Kim, J.; Han, N.; Lee, H.E.; Kim, M.A.; Lee, H.S.; Kim, W.H. Caveolin 1 Expression Correlates with Poor Prognosis and Focal Adhesion Kinase Expression in Gastric Cancer. Pathobiology 2013, 80, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.M.; Liu, J. Caveolin-1 Up-Regulation during Epithelial to Mesenchymal Transition Is Mediated by Focal Adhesion Kinase. J. Biol. Chem. 2008, 283, 13714–13724. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.; Naidoo, M.; Machado, L.R.; Anthony, K. The Duchenne Muscular Dystrophy Gene and Cancer. Cell. Oncol. 2021, 44, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Matissek, S.J.; Elsawa, S.F. GLI3: A Mediator of Genetic Diseases, Development and Cancer. Cell Commun. Signal. 2020, 18, 54. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Song, S.; Wang, Z.; Ajani, J.A. The Role of Hedgehog Signaling in Gastric Cancer: Molecular Mechanisms, Clinical Potential, and Perspective. Cell Commun. Signal. 2019, 17, 157. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ma, L.; Zhang, Z.; Liu, X.; Gao, H.; Zhuang, Y.; Yang, P.; Kornmann, M.; Tian, X.; Yang, Y. Hedgehog Signaling Regulates Epithelial-Mesenchymal Transition in Pancreatic Cancer Stem-Like Cells. J. Cancer 2016, 7, 408–417. [Google Scholar] [CrossRef]
- Fattahi, S.; Nikbakhsh, N.; Ranaei, M.; Sabour, D.; Akhavan-Niaki, H. Association of Sonic Hedgehog Signaling Pathway Genes IHH, BOC, RAB23a and MIR195-5p, MIR509-3-5p, MIR6738-3p with Gastric Cancer Stage. Sci. Rep. 2021, 11, 7471. [Google Scholar] [CrossRef] [PubMed]
- Caparrós-Martín, J.A.; Valencia, M.; Reytor, E.; Pacheco, M.; Fernandez, M.; Perez-Aytes, A.; Gean, E.; Lapunzina, P.; Peters, H.; Goodship, J.A.; et al. The Ciliary Evc/Evc2 Complex Interacts with Smo and Controls Hedgehog Pathway Activity in Chondrocytes by Regulating Sufu/Gli3 Dissociation and Gli3 Trafficking in Primary Cilia. Hum. Mol. Genet. 2013, 22, 124–139. [Google Scholar] [CrossRef]
- Rodrigues, M.F.; Miguita, L.; De Andrade, N.; Heguedusch, D.; Rodini, C.; Moyses, R.; Toporcov, T.; Gama, R.; Tajara, E.; Nunes, F. GLI3 Knockdown Decreases Stemness, Cell Proliferation and Invasion in Oral Squamous Cell Carcinoma. Int. J. Oncol. 2018, 53, 2458–2472. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qiu, M.; An, Y.; Huang, J.; Gong, C. MiR-7-5p Acts as a Tumor Suppressor in Bladder Cancer by Regulating the Hedgehog Pathway Factor Gli3. Biochem. Biophys. Res. Commun. 2018, 503, 2101–2107. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.N.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-Genome Sequencing and Comprehensive Molecular Profiling Identify New Driver Mutations in Gastric Cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Zhang, Z.; Wang, P. GLI3 Promotes Invasion and Predicts Poor Prognosis in Colorectal Cancer. BioMed Res. Int. 2021, 2021, 8889986. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in Targeting Cyclic Nucleotide Phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Gong, J.; Jin, Y.; Zhou, Y.; Tong, R.; Wei, X.; Bai, L.; Shi, J. Inhibitors of Phosphodiesterase as Cancer Therapeutics. Eur. J. Med. Chem. 2018, 150, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Zeng, H.; Li, J.; Xiao, L.; He, Y.; Tang, Y.; Li, Y. MiR-199a Regulates the Tumor Suppressor Mitogen-Activated Protein Kinase Kinase Kinase 11 in Gastric Cancer. Biol. Pharm. Bull. 2010, 33, 1822–1827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fan, K.-J.; Sun, Q.; Chen, A.-Z.; Shen, W.-L.; Zhao, Z.-H.; Zheng, X.-F.; Yang, X. Functional Screening for MiRNAs Targeting Smad4 Identified MiR-199a as a Negative Regulator of TGF-β Signalling Pathway. Nucleic Acids Res. 2012, 40, 9286–9297. [Google Scholar] [CrossRef]
- Yu, L.; Cao, C.; Li, X.; Zhang, M.; Gu, Q.; Gao, H.; Balic, J.J.; Xu, D.; Zhang, L.; Ying, L.; et al. Complete Loss of MiR-200 Family Induces EMT Associated Cellular Senescence in Gastric Cancer. Oncogene 2022, 41, 26–36. [Google Scholar] [CrossRef]
- Liang, Z.; Li, X.; Liu, S.; Li, C.; Wang, X.; Xing, J. MiR-141–3p Inhibits Cell Proliferation, Migration and Invasion by Targeting TRAF5 in Colorectal Cancer. Biochem. Biophys. Res. Commun. 2019, 514, 699–705. [Google Scholar] [CrossRef]
- Huang, M.; Wu, L.; Qin, Y.; Li, Z.; Luo, S.; Qin, H.; Yang, Y.; Chen, J. Anti-Proliferative Role and Prognostic Implication of MiR-141 in Gastric Cancer. Am. J. Transl. Res. 2016, 8, 3549–3557. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort | Subtype | Sample Size | Age (Mean ± sd) | Sex | AJCC Pathologic Stage | ||||
|---|---|---|---|---|---|---|---|---|---|
| Male | Female | I | II | III | IV | ||||
| TCGA-STAD | EMT | 47 | 61.7 ± 10.06 | 62% | 38% | 7% | 33% | 51% | 9% |
| MSI | 37 | 70.16 ± 10.58 | 54% | 46% | 27% | 32% | 30% | 11% | |
| TP53+ | 42 | 66.44 ± 11.02 | 71% | 29% | 15% | 41% | 39% | 5% | |
| TP53− | 41 | 66.92 ± 9.59 | 68% | 32% | 23% | 36% | 31% | 10% | |
| NA | 149 | 66.77 ± 11.02 | 64% | 36% | 13% | 27% | 48% | 12% | |
| ACRG | EMT | 46 | 55.72 ± 12.44 | 59% | 41% | 4% | 15% | 39% | 41% |
| MSI | 68 | 64.82 ± 9.94 | 66% | 34% | 21% | 38% | 28% | 13% | |
| TP53+ | 78 | 61.86 ± 11.67 | 72% | 28% | 5% | 38% | 34% | 23% | |
| TP53− | 105 | 62.86 ± 10.48 | 65% | 35% | 8% | 32% | 31% | 30% | |
| Singapore | EMT | 83 | 62.64 ± 13.15 | 60% | 40% | 11% | 16% | 36% | 37% |
| MSI | 11 | 69.33 ± 12.67 | 55% | 45% | 36% | 9% | 36% | 18% | |
| TP53+ | 37 | 63.15 ± 13.2 | 73% | 27% | 19% | 16% | 38% | 27% | |
| TP53− | 61 | 66.58 ± 13.24 | 69% | 31% | 18% | 15% | 39% | 28% | |
| Node1 | Node2 | Node3 | HR in TCGA | Cox Regression p-Value in TCGA | HR in ACRG | Cox Regression p-Value in ACRG |
|---|---|---|---|---|---|---|
| ACTN2 | LDB3 | PDLIM3 | 1.199 | 0.51 | 2.936 | 0.019 |
| ADCY5 | CAV1 | CACNA1C | 2.396 | 0.007 | 4.406 | >0.001 |
| CAMK2A | ADCY5 | CACNA1C | 1.853 | 0.054 | 1.234 | 0.585 |
| CAMK2A | ACTN1 | CACNA1C | 2.499 | 0.003 | 0.958 | 0.919 |
| CAMK2A | ADCY5 | ADCY2 | 1.716 | 0.077 | 0.994 | 0.989 |
| CNN1 | MYH11 | ACTG2 | 1.608 | 0.042 | 2.179 | >0.001 |
| DMD | ITGA1 | CAV1 | 3.636 | >0.001 | 3.13 | >0.001 |
| EVC | EVC2 | GLI3 | 2.035 | 0.007 | 2.746 | >0.001 |
| FLNA | ITGB3 | CAV1 | 2.997 | 0.001 | 2.088 | 0.019 |
| FLNA | ITGB3 | VCL | 2.438 | 0.01 | 2.299 | 0.03 |
| GUCY1A1 | GUCY1B1 | PDE3A | 1.716 | 0.034 | 1.905 | 0.006 |
| GUCY1A1 | GUCY1B1 | PRKG1 | 1.852 | 0.012 | 1.76 | 0.02 |
| IGF1 | FGF7 | FGFR1 | 2.133 | 0.009 | 2.47 | 0.001 |
| IGF1 | FGF10 | FGFR1 | 1.926 | 0.02 | 2.984 | 0.002 |
| IGF1 | FGF10 | HGF | 2.223 | 0.009 | 1.768 | 0.054 |
| IGF1 | FGF10 | KIT | 1.622 | 0.102 | 1.559 | 0.104 |
| IGF1 | FGF2 | FGFR1 | 1.741 | 0.051 | 2.303 | 0.003 |
| IGF1 | FGF2 | KIT | 1.388 | 0.273 | 1.377 | 0.233 |
| IGF1 | FGF2 | HGF | 1.874 | 0.033 | 1.663 | 0.056 |
| IGF1 | FGF7 | KIT | 1.691 | 0.088 | 1.489 | 0.12 |
| ITGA1 | ITGB3 | CAV1 | 4.165 | >0.001 | 2.079 | 0.009 |
| ITGA9 | JAM3 | JAM2 | 2.101 | 0.004 | 2.13 | 0.003 |
| ITGB3 | VCL | ACTN1 | 2.45 | 0.014 | 1.669 | 0.22 |
| KCNA1 | LGI1 | CNTN2 | 1.184 | 0.542 | 1.113 | 0.782 |
| LMOD1 | MYH11 | ACTG2 | 1.534 | 0.065 | 2.105 | 0.002 |
| LMOD1 | CNN1 | ACTG2 | 1.599 | 0.044 | 1.92 | 0.004 |
| LMOD1 | CNN1 | MYH11 | 1.43 | 0.113 | 1.872 | 0.006 |
| MYH11 | MYL9 | ACTG2 | 2.106 | 0.005 | 3.318 | >0.001 |
| MYH11 | TAGLN | ACTG2 | 1.741 | 0.03 | 2.52 | >0.001 |
| MYLK | MYH11 | ACTG2 | 1.552 | 0.071 | 2.691 | >0.001 |
| MYOCD | CNN1 | MYH11 | 1.487 | 0.096 | 2.002 | 0.003 |
| OGN | OMD | PRELP | 2.052 | 0.005 | 1.48 | 0.095 |
| OGN | ST3GAL3 | OMD | 1.614 | 0.079 | 1.725 | 0.069 |
| OGN | ST3GAL3 | PRELP | 1.797 | 0.037 | 2.089 | 0.017 |
| PDE1A | GUCY1A1 | GUCY1B1 | 1.981 | 0.009 | 1.761 | 0.018 |
| PDE2A | GUCY1A1 | GUCY1B1 | 2.254 | 0.003 | 2.23 | 0.003 |
| PRNP | CAV1 | CACNA1C | 2.972 | 0.003 | 4.006 | >0.001 |
| SNAP25 | CAV1 | CACNA1C | 3.014 | 0.001 | 3.29 | 0.001 |
| TPM2 | MYH11 | ACTG2 | 1.648 | 0.069 | 2.901 | >0.001 |
| Node1 | Node2 | Node3 | AUC in the Training Set (TCGA) | AUC in the Validation Set (ACRG) | AUC in the Independent Set (Singapore) |
|---|---|---|---|---|---|
| EVC | EVC2 | GLI3 | 0.943 | 0.974 | 0.92 |
| PDE2A | GUCY1A1 | GUCY1B1 | 0.935 | 0.972 | 0.947 |
| IGF1 | FGF2 | FGFR1 | 0.932 | 0.969 | 0.935 |
| ITGA9 | JAM3 | JAM2 | 0.944 | 0.969 | 0.935 |
| GUCY1A1 | GUCY1B1 | PDE3A | 0.927 | 0.967 | 0.944 |
| IGF1 | FGF7 | FGFR1 | 0.938 | 0.967 | 0.926 |
| GUCY1A1 | GUCY1B1 | PRKG1 | 0.927 | 0.965 | 0.944 |
| IGF1 | FGF10 | FGFR1 | 0.941 | 0.965 | 0.932 |
| SNAP25 | CAV1 | CACNA1C | 0.877 | 0.961 | 0.904 |
| PRNP | CAV1 | CACNA1C | 0.9 | 0.954 | 0.914 |
| MYLK | MYH11 | ACTG2 | 0.908 | 0.952 | 0.913 |
| PDE1A | GUCY1A1 | GUCY1B1 | 0.936 | 0.949 | 0.941 |
| ACTN2 | LDB3 | PDLIM3 | 0.927 | 0.948 | 0.934 |
| IGF1 | FGF2 | KIT | 0.91 | 0.944 | 0.9 |
| IGF1 | FGF2 | HGF | 0.911 | 0.942 | 0.907 |
| ADCY5 | CAV1 | CACNA1C | 0.893 | 0.939 | 0.892 |
| IGF1 | FGF7 | KIT | 0.923 | 0.937 | 0.902 |
| MYH11 | MYL9 | ACTG2 | 0.904 | 0.937 | 0.916 |
| MYH11 | TAGLN | ACTG2 | 0.915 | 0.935 | 0.918 |
| DMD | ITGA1 | CAV1 | 0.883 | 0.929 | 0.899 |
| OGN | OMD | PRELP | 0.935 | 0.925 | 0.914 |
| FLNA | ITGB3 | VCL | 0.888 | 0.921 | 0.92 |
| OGN | ST3GAL3 | OMD | 0.937 | 0.92 | 0.893 |
| IGF1 | FGF10 | KIT | 0.931 | 0.918 | 0.904 |
| OGN | ST3GAL3 | PRELP | 0.938 | 0.916 | 0.883 |
| FLNA | ITGB3 | CAV1 | 0.897 | 0.915 | 0.925 |
| IGF1 | FGF10 | HGF | 0.928 | 0.915 | 0.911 |
| LMOD1 | CNN1 | ACTG2 | 0.899 | 0.912 | 0.876 |
| CNN1 | MYH11 | ACTG2 | 0.889 | 0.911 | 0.836 |
| ITGA1 | ITGB3 | CAV1 | 0.876 | 0.906 | 0.904 |
| TPM2 | MYH11 | ACTG2 | 0.864 | 0.882 | 0.881 |
| ITGB3 | VCL | ACTN1 | 0.815 | 0.881 | 0.868 |
| LMOD1 | MYH11 | ACTG2 | 0.897 | 0.877 | 0.88 |
| LMOD1 | CNN1 | MYH11 | 0.887 | 0.876 | 0.886 |
| CAMK2A | ACTN1 | CACNA1C | 0.888 | 0.864 | 0.814 |
| MYOCD | CNN1 | MYH11 | 0.847 | 0.829 | 0.844 |
| KCNA1 | LGI1 | CNTN2 | 0.914 | 0.783 | 0.587 |
| CAMK2A | ADCY5 | ADCY2 | 0.887 | 0.777 | 0.732 |
| CAMK2A | ADCY5 | CACNA1C | 0.895 | 0.765 | 0.725 |
| Node1 | Node2 | Node3 | AUC in the TCGA | AUC in the GSE184336 (Training) | AUC in the GSE184336 (Validation) |
|---|---|---|---|---|---|
| PDE2A | GUCY1A1 | GUCY1B1 | 0.95 | 0.772 | 0.854 |
| DMD | ITGA1 | CAV1 | 0.932 | 0.835 | 0.822 |
| KCNA1 | LGI1 | CNTN2 | 0.929 | 0.83 | 0.826 |
| PDE1A | GUCY1A1 | GUCY1B1 | 0.92 | 0.737 | 0.803 |
| MYLK | MYH11 | ACTG2 | 0.914 | 0.821 | 0.884 |
| ITGA9 | JAM3 | JAM2 | 0.912 | 0.663 | 0.614 |
| ADCY5 | CAV1 | CACNA1C | 0.904 | 0.696 | 0.804 |
| IGF1 | FGF7 | KIT | 0.895 | 0.888 | 0.868 |
| IGF1 | FGF2 | KIT | 0.893 | 0.888 | 0.868 |
| IGF1 | FGF10 | KIT | 0.889 | 0.887 | 0.864 |
| ITGA1 | ITGB3 | CAV1 | 0.876 | 0.699 | 0.757 |
| CAMK2A | ADCY5 | CACNA1C | 0.858 | 0.752 | 0.807 |
| OGN | OMD | PRELP | 0.857 | 0.629 | 0.691 |
| ACTN2 | LDB3 | PDLIM3 | 0.85 | 0.74 | 0.709 |
| LMOD1 | CNN1 | MYH11 | 0.85 | 0.707 | 0.705 |
| OGN | ST3GAL3 | OMD | 0.849 | 0.615 | 0.7 |
| LMOD1 | MYH11 | ACTG2 | 0.844 | 0.682 | 0.707 |
| LMOD1 | CNN1 | ACTG2 | 0.844 | 0.689 | 0.677 |
| IGF1 | FGF2 | HGF | 0.831 | 0.843 | 0.83 |
| TPM2 | MYH11 | ACTG2 | 0.829 | 0.704 | 0.72 |
| FLNA | ITGB3 | CAV1 | 0.828 | 0.691 | 0.728 |
| MYH11 | TAGLN | ACTG2 | 0.822 | 0.787 | 0.812 |
| IGF1 | FGF10 | HGF | 0.821 | 0.813 | 0.817 |
| CNN1 | MYH11 | ACTG2 | 0.82 | 0.702 | 0.701 |
| MYOCD | CNN1 | MYH11 | 0.82 | 0.698 | 0.689 |
| PRNP | CAV1 | CACNA1C | 0.815 | 0.634 | 0.69 |
| MYH11 | MYL9 | ACTG2 | 0.814 | 0.751 | 0.754 |
| SNAP25 | CAV1 | CACNA1C | 0.813 | 0.74 | 0.832 |
| OGN | ST3GAL3 | PRELP | 0.808 | 0.58 | 0.606 |
| GUCY1A1 | GUCY1B1 | PRKG1 | 0.807 | 0.741 | 0.783 |
| CAMK2A | ADCY5 | ADCY2 | 0.792 | 0.646 | 0.566 |
| FLNA | ITGB3 | VCL | 0.768 | 0.687 | 0.735 |
| IGF1 | FGF2 | FGFR1 | 0.762 | 0.828 | 0.776 |
| IGF1 | FGF10 | FGFR1 | 0.752 | 0.773 | 0.736 |
| CAMK2A | ACTN1 | CACNA1C | 0.745 | 0.802 | 0.811 |
| EVC | EVC2 | GLI3 | 0.734 | 0.658 | 0.567 |
| GUCY1A1 | GUCY1B1 | PDE3A | 0.665 | 0.718 | 0.748 |
| IGF1 | FGF7 | FGFR1 | 0.655 | 0.791 | 0.777 |
| ITGB3 | VCL | ACTN1 | 0.613 | 0.712 | 0.703 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadeghi, M.; Karimi, M.R.; Karimi, A.H.; Ghorbanpour Farshbaf, N.; Barzegar, A.; Schmitz, U. Network-Based and Machine-Learning Approaches Identify Diagnostic and Prognostic Models for EMT-Type Gastric Tumors. Genes 2023, 14, 750. https://doi.org/10.3390/genes14030750
Sadeghi M, Karimi MR, Karimi AH, Ghorbanpour Farshbaf N, Barzegar A, Schmitz U. Network-Based and Machine-Learning Approaches Identify Diagnostic and Prognostic Models for EMT-Type Gastric Tumors. Genes. 2023; 14(3):750. https://doi.org/10.3390/genes14030750
Chicago/Turabian StyleSadeghi, Mehdi, Mohammad Reza Karimi, Amir Hossein Karimi, Nafiseh Ghorbanpour Farshbaf, Abolfazl Barzegar, and Ulf Schmitz. 2023. "Network-Based and Machine-Learning Approaches Identify Diagnostic and Prognostic Models for EMT-Type Gastric Tumors" Genes 14, no. 3: 750. https://doi.org/10.3390/genes14030750
APA StyleSadeghi, M., Karimi, M. R., Karimi, A. H., Ghorbanpour Farshbaf, N., Barzegar, A., & Schmitz, U. (2023). Network-Based and Machine-Learning Approaches Identify Diagnostic and Prognostic Models for EMT-Type Gastric Tumors. Genes, 14(3), 750. https://doi.org/10.3390/genes14030750