
Hereditary Tyrosinemia Type 1 Mice under Continuous Nitisinone Treatment Display Remnants of an Uncorrected Liver Disease Phenotype
 , , , , , , and
, , , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
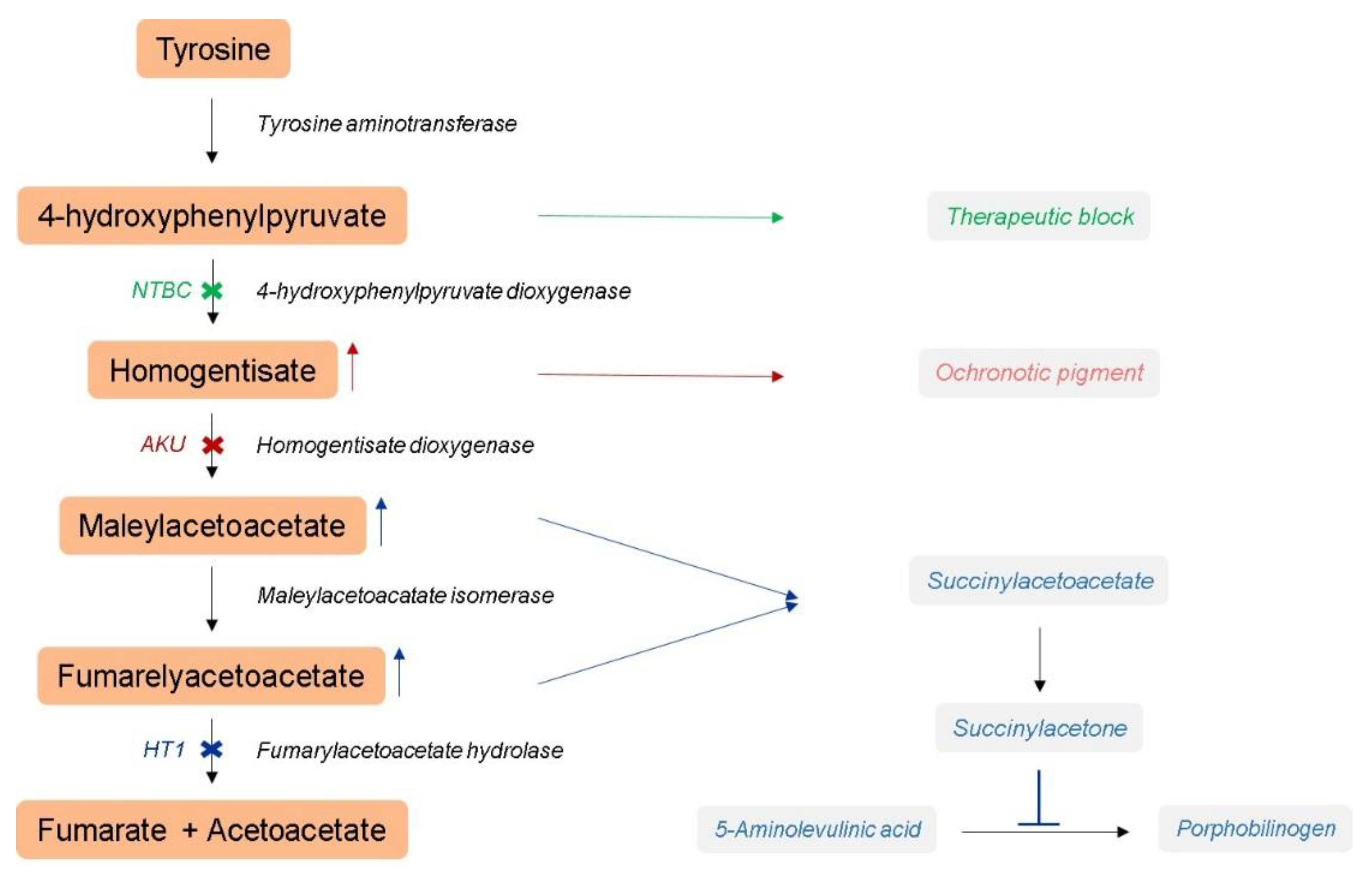
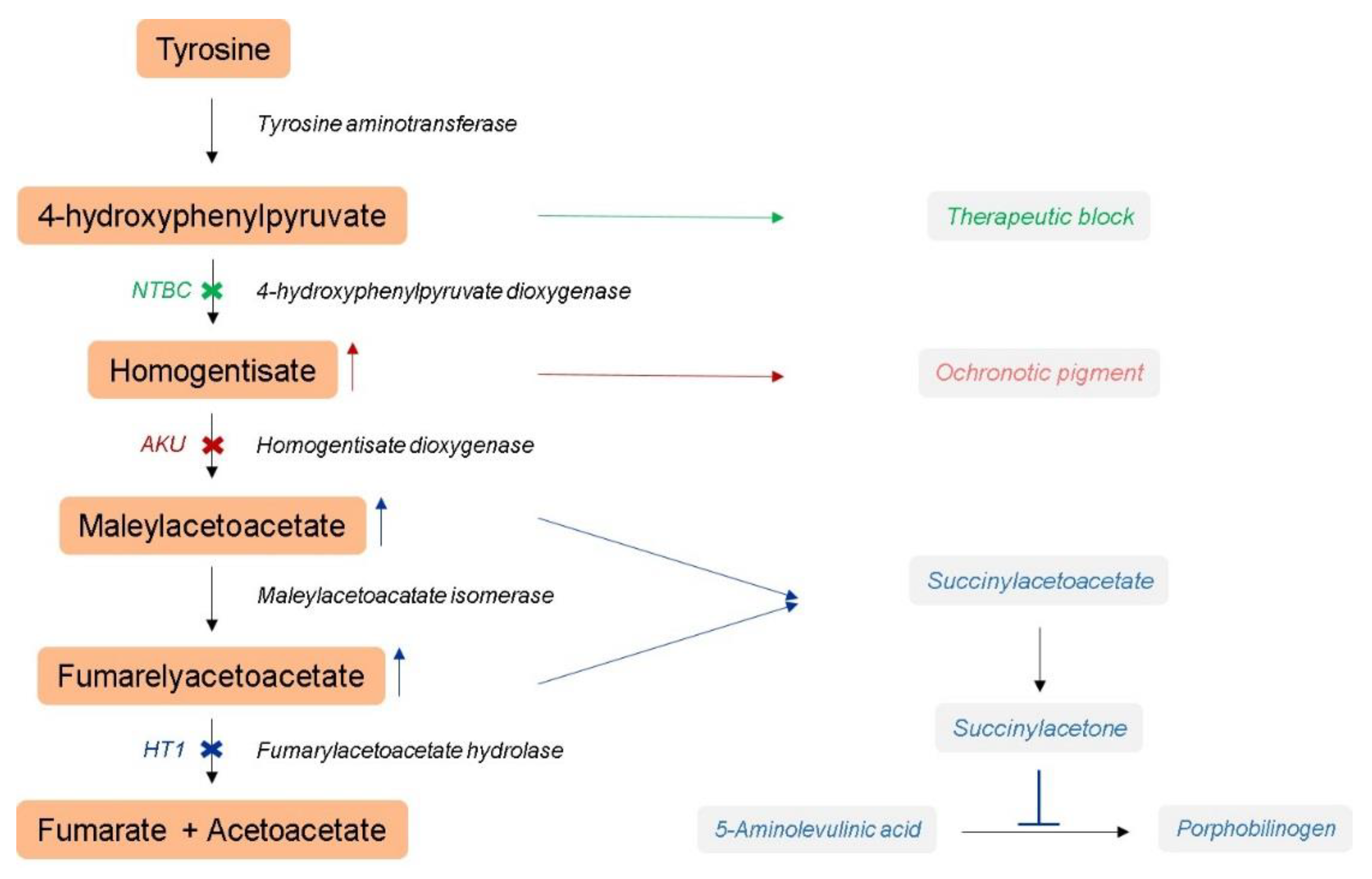
:1. Introduction
2. Materials and Methods
2.1. Hereditary Tyrosinemia Type 1 Mouse Model
2.2. Alkaptonuria Mouse Model
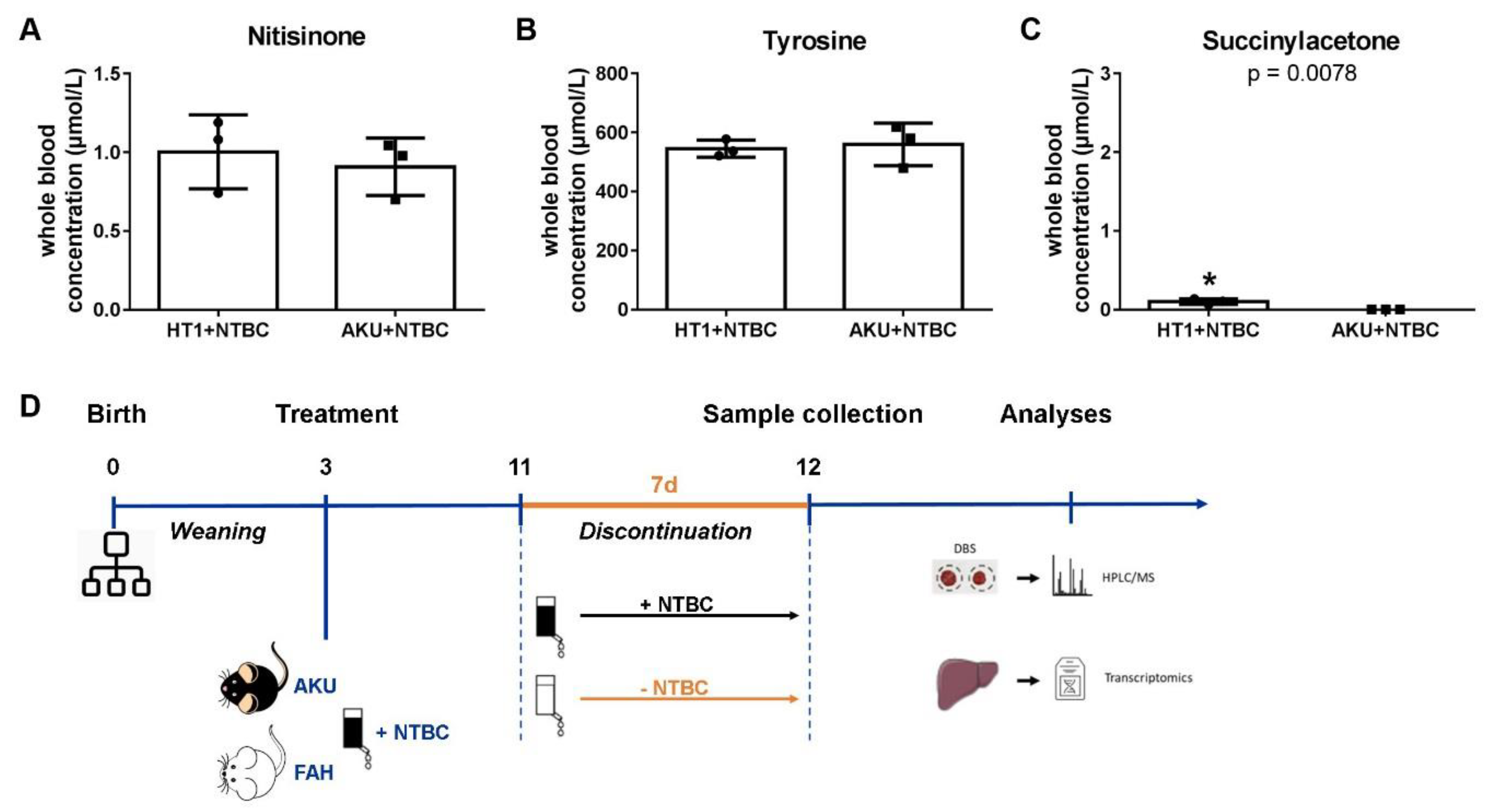
2.3. Animal Experiments
2.4. Sample Collection and Preparation
2.5. Dried Blood Spot Analysis
2.6. Extraction of Total RNA
2.7. Microarray Profiling and Analysis
3. Results
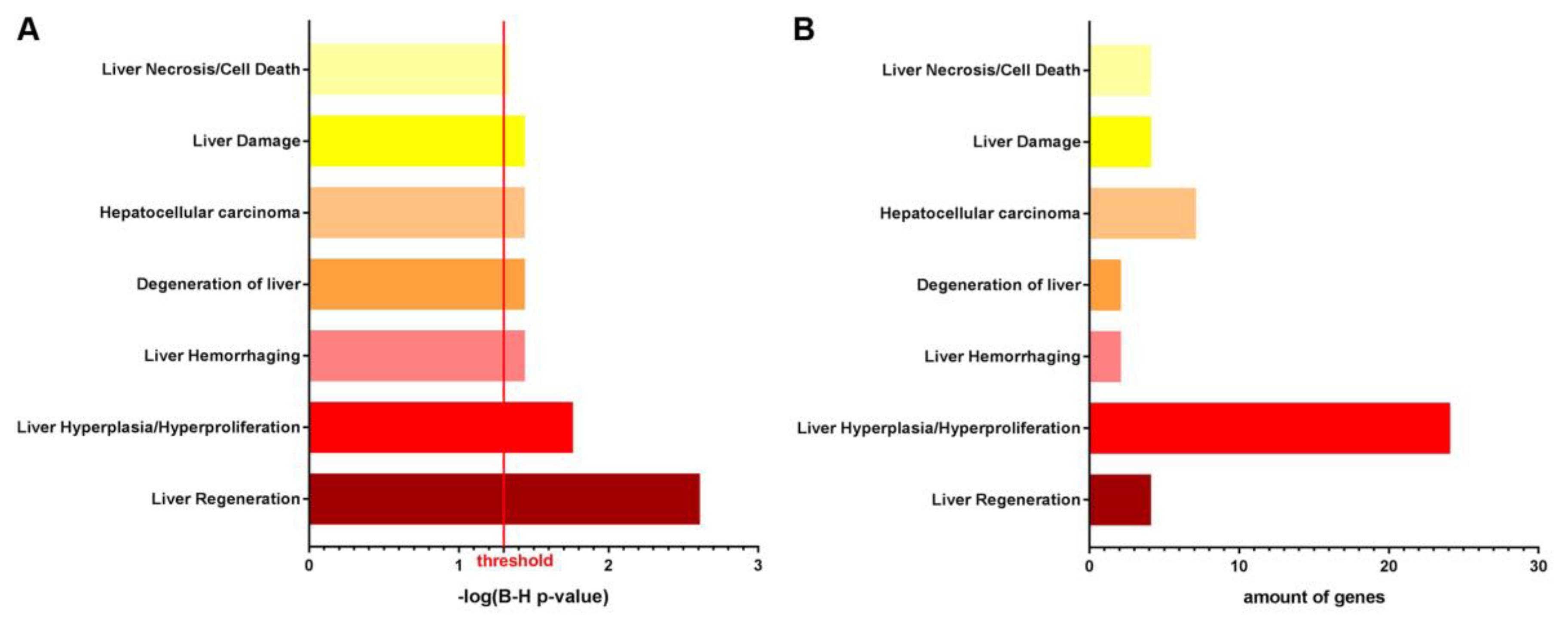
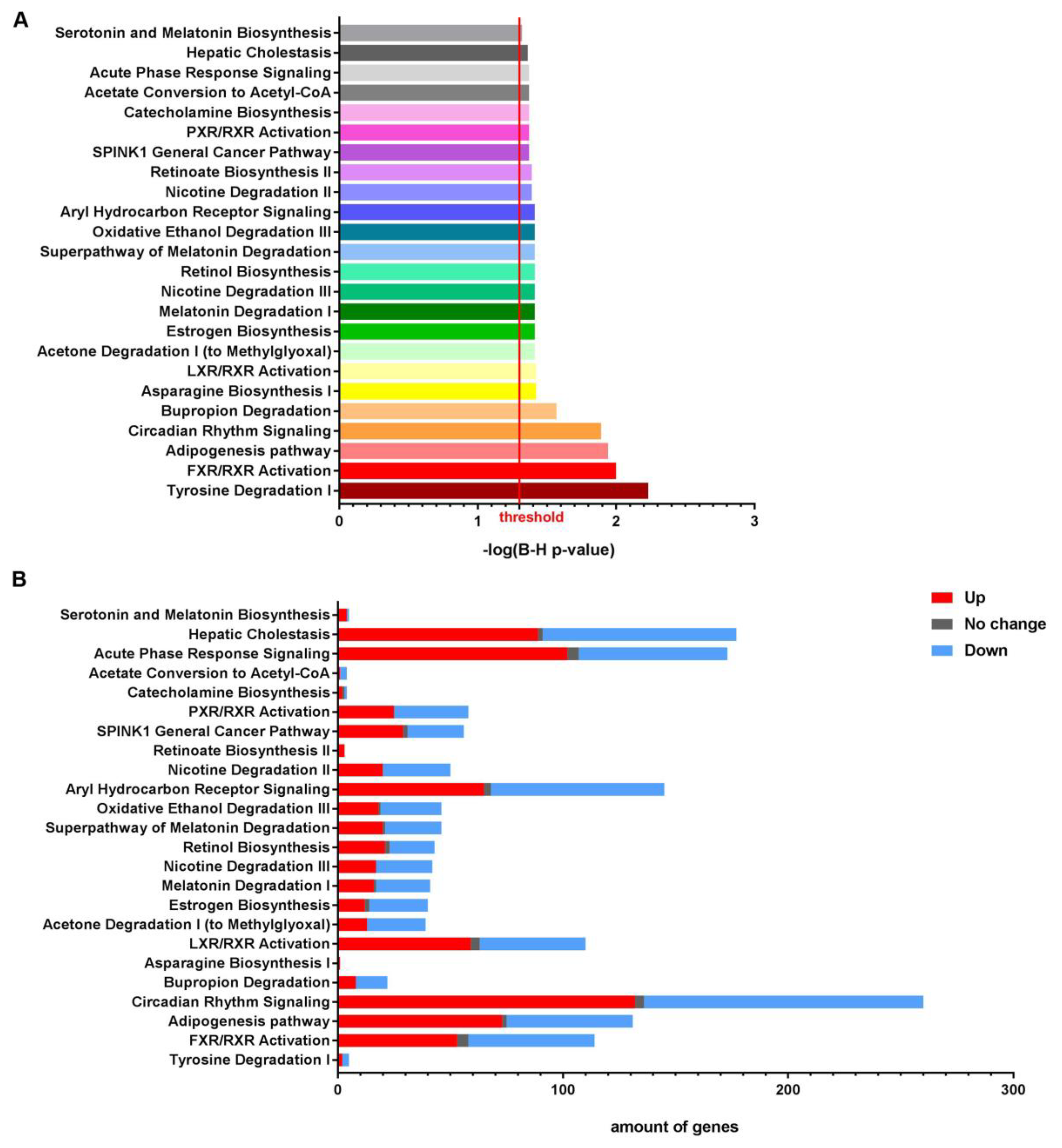
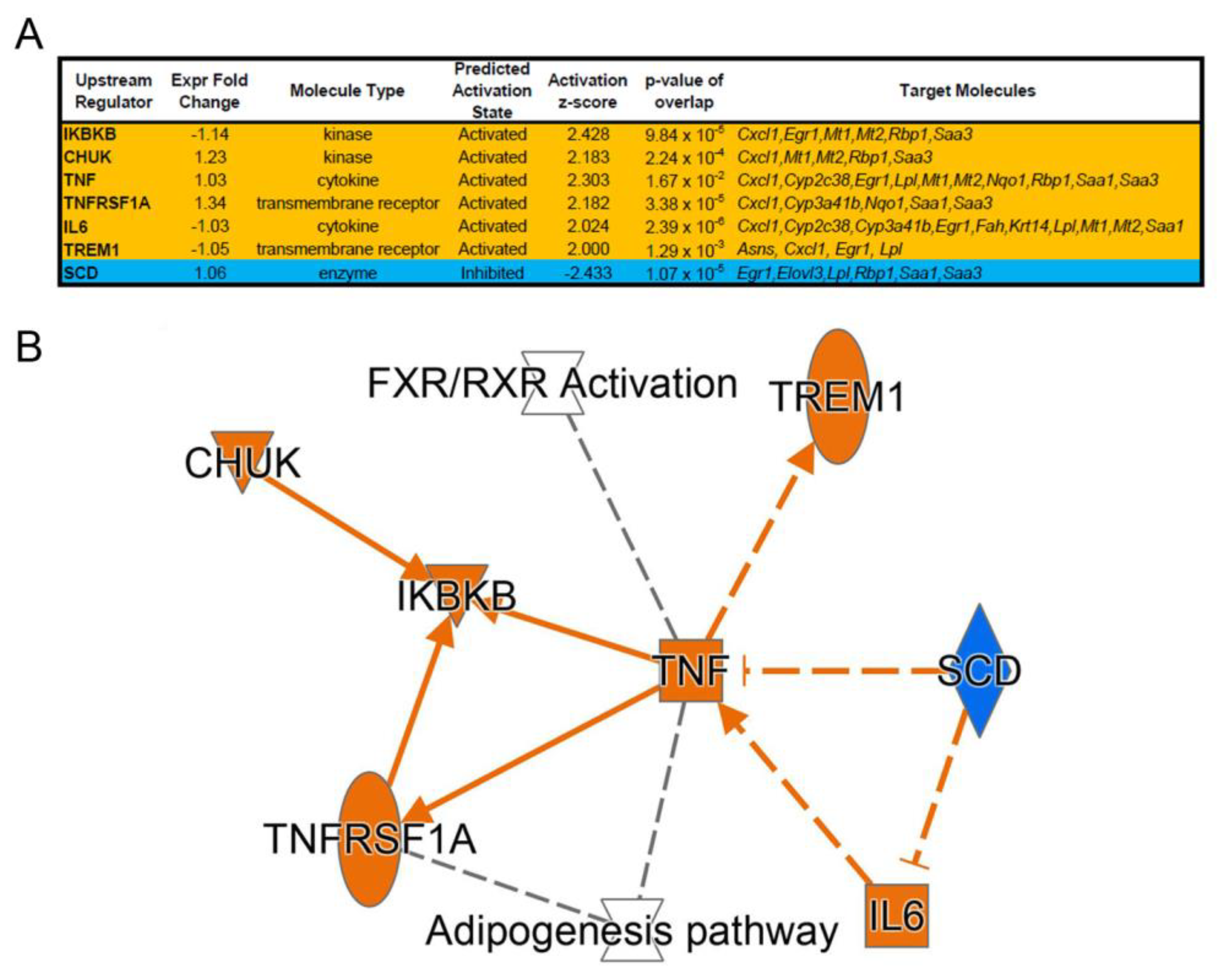
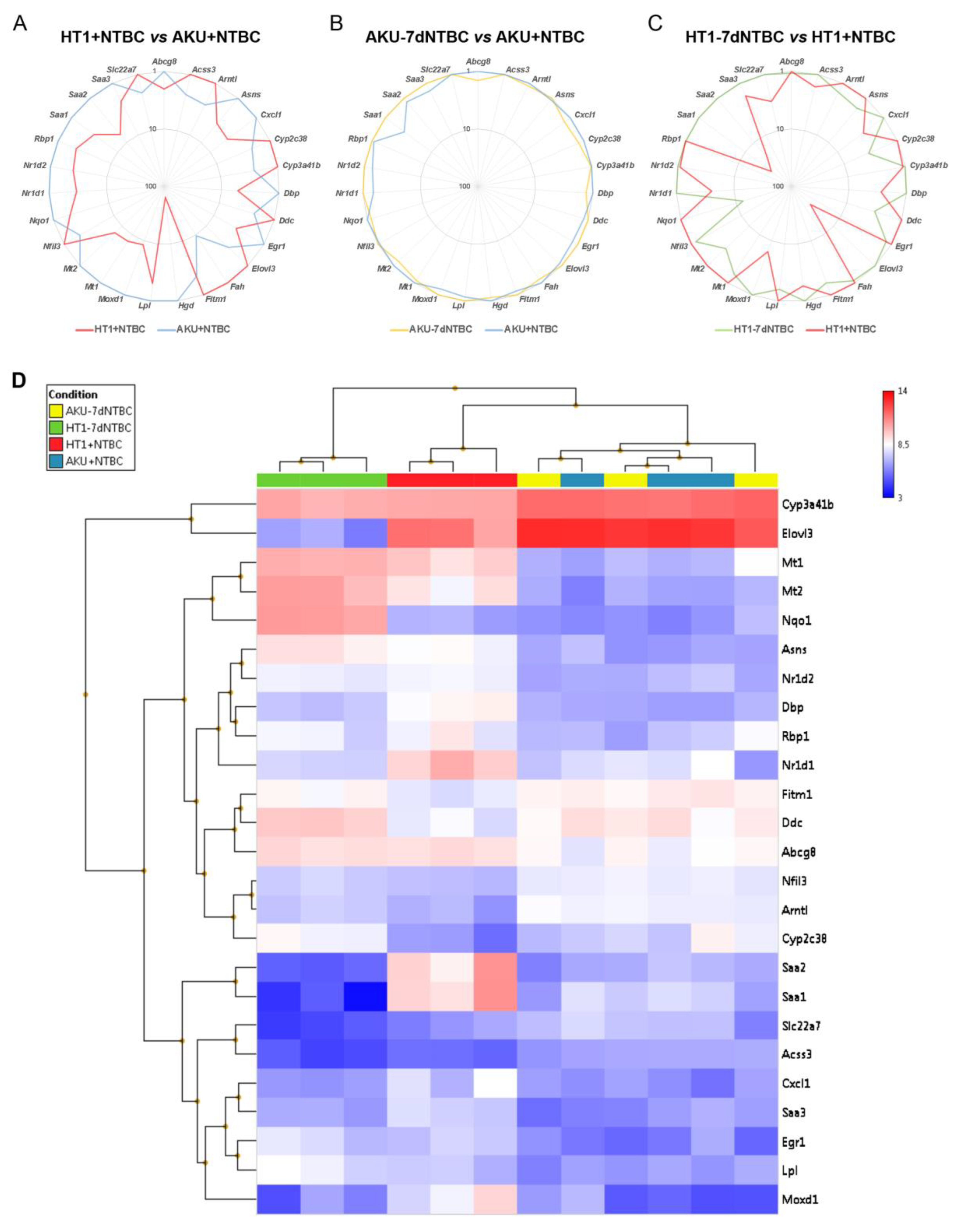
3.1. NTBC Treated Fah-Deficient Mice Show Affected Molecular Pathways Involved in Liver-Specific Metabolic Processes, Lipid Homeostasis and Hepatic Cholestasis Compared to NTBC-Treated Hgd-Deficient Mice
3.2. mRNA Marker Profile of the Uncorrected Liver Disease Phenotype in HT1 vs. AKU Liver and Differential Impact of NTBC Discontinuation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yudkoff, M. Disorders of Amino Acid Metabolism. In Basic Neurochemistry, Principles of Molecular, Cellular, and Medical Neurobiology, 8 ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 737–754. [Google Scholar]
- Morrow, G.; Tanguay, R.M. Biochemical and clinical aspects of hereditary tyrosinemia type 1. Adv. Exp. Med. Biol. 2017, 959, 9–21. [Google Scholar] [PubMed]
- Chakrapani, A.; Gissen, P.; McKiernan, P. Disorders of tyrosine metabolism. In Inborn Metabolic Diseases: Diagnosis and Treatmen; Springer: Berlin/Heidelberg, Germany, 2012; pp. 265–276. [Google Scholar]
- de Laet, C.; Dionisi-Vici, C.; Leonard, J.V.; McKiernan, P.; Mitchell, G.; Monti, L.; de Baulny, H.O.; Pintos-Morell, G.; Spiekerkötter, U. Recommendations for the management of tyrosinaemia type 1. Orphanet J. Rare Dis. 2013, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Äärelä, L.; Hiltunen, P.; Soini, T.; Vuorela, N.; Huhtala, H.; Nevalainen, P.I.; Heikinheimo, M.; Kivelä, L.; Kurppa, K. Type 1 tyrosinemia in Finland: A nationwide study. Orphanet J. Rare Dis. 2020, 15, 1–11. [Google Scholar] [CrossRef]
- Bliksrud, Y.T.; Brodtkorb, E.; Backe, P.H.; Woldseth, B.; Rootwelt, H. Hereditary tyrosinaemia type i in Norway: Incidence and three novel small deletions in the fumarylacetoacetase gene. Scand. J. Clin. Lab. Investig. 2012, 72, 369–373. [Google Scholar] [CrossRef]
- Chinsky, J.M.; Singh, R.; Ficicioglu, C.; van Karnebeek, C.D.M.; Grompe, M.; Mitchell, G.; Waisbren, S.E.; Gucsavas-Calikoglu, M.; Wasserstein, M.P.; Coakley, K.; et al. Diagnosis and treatment of tyrosinemia type I: A US and Canadian consensus group review and recommendations. Genet. Med. 2017, 19, 1380–1395. [Google Scholar] [CrossRef] [Green Version]
- Russo, P.A.; Mitchell, G.A.; Tanguay, R.M. Tyrosinemia: A review. Pediatr. Dev. Pathol. 2001, 4, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Saudubray, J.M.; Van Den Berghe, G.; Walter, J.H. Inborn metabolic diseases diagnosis and treatment. In Inborn Metabolic Diseases: Diagnosis and Treatment; Springer Science & Business Media: Berlin, Germany, 2012; Volume 660, pp. 1–657. [Google Scholar]
- van Ginkel, W.G.; Rodenburg, I.L.; Harding, C.O.; Hollak, C.E.M.; Heiner-Fokkema, M.R.; van Spronsen, F.J. Long-Term Outcomes and Practical Considerations in the Pharmacological Management of Tyrosinemia Type 1. Pediatr. Drugs 2019, 21, 413–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.M. Clinical utility of nitisinone for the treatment of hereditary tyrosinemia type-1 (HT-1). Appl. Clin. Genet. 2017, 10, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Ranganath, L.R.; Milan, A.M.; Hughes, A.T.; Dutton, J.J.; Fitzgerald, R.; Briggs, M.C.; Bygott, H.; Psarelli, E.E.; Cox, T.F.; Gallagher, J.A.; et al. Suitability of nitisinone In alkaptonuria 1 (SONIA 1): An international, multicentre, randomised, open-label, no-treatment controlled, parallel-group, dose-response study to investigate the effect of once daily nitisinone on 24-h urinary homogentisic acid. Ann. Rheum. Dis. 2016, 75, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Ranganath, L.R.; Jarvis, J.C.; Gallagher, J.A. Recent advances in management of alkaptonuria. J. Clin. Pathol. 2013, 66, 367–373. [Google Scholar] [CrossRef]
- Keenan, C.M.; Preston, A.J.; Sutherland, H.; Wilson, P.J.; Psarelli, E.E.; Cox, T.F.; Ranganath, L.R.; Jarvis, J.C.; Gallagher, J.A. Nitisinone arrests but does not reverse ochronosis in alkaptonuric mice. JIMD Rep. 2015, 24, 45–50. [Google Scholar] [PubMed] [Green Version]
- Larochelle, J.; Alvarez, F.; Bussières, J.F.; Chevalier, I.; Dallaire, L.; Dubois, J.; Faucher, F.; Fenyves, D.; Goodyer, P.; Grenier, A.; et al. Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in Québec. Mol. Genet. Metab. 2012, 107, 49–54. [Google Scholar] [CrossRef]
- Gertsman, I.; Gangoiti, J.A.; Nyhan, W.L.; Barshop, B.A. Perturbations of tyrosine metabolism promote the indolepyruvate pathway via tryptophan in host and microbiome. Mol. Genet. Metab. 2015, 114, 431–437. [Google Scholar] [CrossRef]
- Gertsman, I.; Barshop, B.A.; Panyard-Davis, J.; Gangoiti, J.A.; Nyhan, W.L. Metabolic effects of increasing doses of nitisinone in the treatment of alkaptonuria. JIMD Rep. 2015, 24, 13–20. [Google Scholar]
- Thimm, E.; Herebian, D.; Assmann, B.; Klee, D.; Mayatepek, E.; Spiekerkoetter, U. Increase of CSF tyrosine and impaired serotonin turnover in tyrosinemia type I. Mol. Genet. Metab. 2011, 102, 122–125. [Google Scholar] [CrossRef]
- García, M.I.; de la Parra, A.; Arias, C.; Arredondo, M.; Cabello, J.F. Long-term cognitive functioning in individuals with tyrosinemia type 1 treated with nitisinone and protein-restricted diet. Mol. Genet. Metab. Rep. 2017, 11, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Van Ginkel, W.G.; Jahja, R.; Huijbregts, S.C.J.; Daly, A.; MacDonald, A.; De Laet, C.; Cassiman, D.; Eyskens, F.; Körver-Keularts, I.M.L.W.; Goyens, P.J.; et al. Neurocognitive outcome in tyrosinemia type 1 patients compared to healthy controls. Orphanet J. Rare Dis. 2016, 11, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ginkel, W.G.; Van Vliet, D.; Burgerhof, J.G.M.; De Blaauw, P.; Rubio Gozalbo, M.E.; Heiner-Fokkema, M.R.; Van Spronsen, F.J. Presumptive brain influx of large neutral amino acids and the effect of phenylalanine supplementation in patients with Tyrosinemia type 1. PLoS ONE 2017, 12, e0185342. [Google Scholar] [CrossRef]
- Lequeue, S.; Neuckermans, J.; Nulmans, I.; Schwaneberg, U.; Vanhaecke, T.; De Kock, J. A robust bacterial high-throughput screening system to evaluate single nucleotide polymorphisms of human homogentisate 1,2-dioxygenase in the context of alkaptonuria. Sci. Rep. 2022, 12, 19452. [Google Scholar] [CrossRef]
- van Ginkel, W.G.; van Vliet, D.; van der Goot, E.; Faassen, M.H.J.R.; Vogel, A.; Heiner-Fokkema, M.R.; van der Zee, E.A.; van Spronsen, F.J. Blood and brain biochemistry and behaviour in NTBC and dietary treated tyrosinemia type 1 mice. Nutrients 2019, 11, 2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zatkova, A.; Ranganath, L.; Kadasi, L. Alkaptonuria: Current Perspectives. Appl. Clin. Genet. 2020, 13, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holme, E.; Lindstedt, S. Nontransplant treatment of tyrosinemia. Clin. Liver Dis. 2000, 4, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Angileri, F.; Roy, V.; Morrow, G.; Scoazec, J.Y.; Gadot, N.; Orejuela, D.; Tanguay, R.M. Molecular changes associated with chronic liver damage and neoplastic lesions in a murine model of hereditary tyrosinemia type 1. Biochim. Biophys. Acta-Mol. Basis Dis. 2015, 1852, 2603–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, J.H.; Liu, K.; Plagge, A.; Wilson, P.J.M.; Sutherland, H.; Norman, B.P.; Hughes, A.T.; Keenan, C.M.; Milan, A.M.; Sakai, T.; et al. Conditional targeting in mice reveals that hepatic homogentisate 1,2-dioxygenase activity is essential in reducing circulating homogentisic acid and for effective therapy in the genetic disease alkaptonuria. Hum. Mol. Genet. 2019, 28, 3928–3939. [Google Scholar] [CrossRef]
- Neuckermans, J.; Mertens, A.; De Win, D.; Schwaneberg, U.; De Kock, J. A robust bacterial assay for high-throughput screening of human 4-hydroxyphenylpyruvate dioxygenase inhibitors. Sci. Rep. 2019, 9, 14145. [Google Scholar] [CrossRef] [Green Version]
- Lock, E.; Ranganath, L.R.; Timmis, O. The Role of Nitisinone in Tyrosine Pathway Disorders. Curr. Rheumatol. Rep. 2014, 16, 457. [Google Scholar] [CrossRef]
- Helliwell, T.R.; Gallagher, J.A.; Ranganath, L. Alkaptonuria—A review of surgical and autopsy pathology. Histopathology 2008, 53, 503–512. [Google Scholar] [CrossRef]
- Nemethova, M.; Radvanszky, J.; Kadasi, L.; Ascher, D.B.; Pires, D.E.V.; Blundell, T.L.; Porfirio, B.; Mannoni, A.; Santucci, A.; Milucci, L.; et al. Twelve novel HGD gene variants identified in 99 alkaptonuria patients: Focus on “black bone disease” in Italy. Eur. J. Hum. Genet. 2016, 24, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Paulk, N.K.; Wursthorn, K.; Wang, Z.; Finegold, M.J.; Kay, M.A.; Grompe, M. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology 2010, 51, 1200–1208. [Google Scholar] [CrossRef] [Green Version]
- Culiat, C.T.; Klebig, M.L.; Liu, Z.; Monroe, H.; Stanford, B.; Desai, J.; Tandan, S.; Hughes, L.; Kerley, M.K.; Carpenter, D.A.; et al. Identification of mutations from phenotype-driven ENU mutagenesis in mouse Chromosome 7. Mamm. Genome 2005, 16, 555–566. [Google Scholar] [CrossRef]
- Colemonts-Vroninks, H.; Norman, P.B.; Van Laere, S.; Davison, S.A.; Marcélis, L.; Casimir, G.; Goyens, P.; Claes, P.; De Bundel, D.; Martens, G.; et al. Short-term nitisinone discontinuation of hereditary tyrosinemia type 1 mice causes metabolic alterations in glutathione metabolism/biosynthesis and multiple amino acid degradation pathways. Genes Dis. 2022, in press.
- Al-Dhalimy, M.; Overturf, K.; Finegold, M.; Grompe, M. Long-term therapy with NTBC and tyrosine-restricted diet in a murine model of hereditary tyrosinemia type I. Mol. Genet. Metab. 2002, 75, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, V.E.S.; Dutra, F.; Soares, C.O.; Alves, A.N.L.; Bevilacqua, E.; Gagioti, S.M.; Penatti, C.A.A.; Bechara, E.J.H. Liver damage induced by succinylacetone: A shared redox imbalance mechanism between tyrosinemia and hepatic porphyrias. J. Braz. Chem. Soc. 2017, 28, 1297–1307. [Google Scholar] [CrossRef] [Green Version]
- You, K.; Wang, Y.; Chen, X.; Yang, Z.; Chen, Y.; Tan, S.; Tao, J.; Getachew, A.; Pan, T.; Xu, Y.; et al. Neutralizing serum amyloid a protects against sinusoidal endothelial cell damage and platelet aggregation during acetaminophen-induced liver injury. Biochem. Biophys. Res. Commun. 2023, 639, 20–28. [Google Scholar] [CrossRef]
- Zhang, B.; Dong, L.W.; Tan, Y.X.; Zhang, J.; Pan, Y.F.; Yang, C.; Li, M.H.; Ding, Z.W.; Liu, L.J.; Jiang, T.Y.; et al. Asparagine synthetase is an independent predictor of surgical survival and a potential therapeutic target in hepatocellular carcinoma. Br. J. Cancer 2013, 109, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lu, Y.F.; Chen, H.; Liu, J. Dysregulation of metallothionein and circadian genes in human hepatocellular carcinoma. Chronobiol. Int. 2017, 34, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Si, M.; Lang, J. The roles of metallothioneins in carcinogenesis. J. Hematol. Oncol. 2018, 11, 107. [Google Scholar] [CrossRef]
- Ozen, E.; Gozukizil, A.; Erdal, E.; Uren, A.; Bottaro, D.P.; Atabey, N. Heparin inhibits Hepatocyte growth factor induced motility and invasion of hepatocellular carcinoma cells through early growth response protein 1. PLoS ONE 2012, 7, e42717. [Google Scholar] [CrossRef] [Green Version]
- Shi, P.; Xu, J.; Xia, F.; Wang, Y.; Ren, J.; Liang, P.; Cui, H. MOXD1 knockdown suppresses the proliferation and tumor growth of glioblastoma cells via ER stress-inducing apoptosis. Cell Death Discov. 2022, 8, 174. [Google Scholar] [CrossRef]
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, Y.; Han, A.; Tang, C.; Xu, R.; Feng, L.; Yang, Y.; Chen, L.; Lin, Z. The NQO1/p53/SREBP1 axis promotes hepatocellular carcinoma progression and metastasis by regulating Snail stability. Oncogene 2022, 41, 5107–5120. [Google Scholar] [CrossRef]
- Zhao, W.; Jiang, L.; Fang, T.; Fang, F.; Liu, Y.; Zhao, Y.; You, Y.; Zhou, H.; Su, X.; Wang, J.; et al. β-Lapachone Selectively Kills Hepatocellular Carcinoma Cells by Targeting NQO1 to Induce Extensive DNA Damage and PARP1 Hyperactivation. Front. Oncol. 2021, 11, 747282. [Google Scholar] [CrossRef]
- Lin, L.; Sun, J.; Tan, Y.; Li, Z.; Kong, F.; Shen, Y.; Liu, C.; Chen, L. Prognostic implication of NQO1 overexpression in hepatocellular carcinoma. Hum. Pathol. 2017, 69, 31–37. [Google Scholar] [CrossRef]
- Dimri, M.; Humphries, A.; Laknaur, A.; Elattar, S.; Lee, T.J.; Sharma, A.; Kolhe, R.; Satyanarayana, A. NAD(P)H Quinone Dehydrogenase 1 Ablation Inhibits Activation of the Phosphoinositide 3-Kinase/Akt Serine/Threonine Kinase and Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Pathways and Blocks Metabolic Adaptation in Hepatocellular. Hepatology 2020, 71, 549–568. [Google Scholar] [CrossRef]
- Krawczyk, M.; Niewiadomska, O.; Jankowska, I.; Jankowski, K.; Więckowski, S.; Lebensztejn, D.; Więcek, S.; Gozdowska, J.; Kułaga, Z.; Weber, S.N.; et al. Common variant p.D19H of the hepatobiliary sterol transporter ABCG8 increases the risk of gallstones in children. Liver Int. 2022, 42, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Lepreux, S.; Bioulac-Sage, P.; Gabbiani, G.; Sapin, V.; Housset, C.; Rosenbaum, J.; Balabaud, C.; Desmoulière, A. Cellular retinol-binding protein-1 expression in normal and fibrotic/cirrhotic human liver: Different patterns of expression in hepatic stellate cells and (myo)fibroblast subpopulations. J. Hepatol. 2004, 40, 774–780. [Google Scholar] [CrossRef]
- Kobayashi, Y. The role of chemokines in neutrophil biology. Front. Biosci. 2008, 13, 2400–2407. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Li, Z.; Gao, J.; Gao, P.J.; Ni, Y.B.; Zhu, J.Y. Elevated CXCL1 increases hepatocellular carcinoma aggressiveness and is inhibited by miRNA-200a. Oncotarget 2016, 7, 65052–65066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiri, K.I.; Richmond, A. Fine Tuning the Transcriptional Regulation of the CXCL1 Chemokine. In Progress in Nucleic Acid Research and Molecular Biology; 2003; Volume 74, pp. 1–36. ISBN 0125400748. [Google Scholar]
- Bertola, A.; Bonnafous, S.; Anty, R.; Patouraux, S.; Saint-Paul, M.C.; Iannelli, A.; Gugenheim, J.; Barr, J.; Mato, J.M.; Le Marchand-Brustel, Y.; et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and nash in morbidly obese patients. PLoS ONE 2010, 5, e13577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, B.; Xu, M.J.; Zhou, Z.; Cai, Y.; Li, M.; Wang, W.; Feng, D.; Bertola, A.; Wang, H.; Kunos, G.; et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: An important role for CXCL1. Hepatology 2015, 62, 1070–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Miura, K.; Zhang, B.; Matsushita, H.; Yang, Y.M.; Liang, S.; Song, J.; Roh, Y.S.; Seki, E. TRIF Differentially Regulates Hepatic Steatosis and Inflammation/Fibrosis in Mice. Cmgh 2017, 3, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.; He, Y.; Xiang, X.; Seo, W.; Kim, S.J.; Ma, J.; Ren, T.; Park, S.H.; Zhou, Z.; Feng, D.; et al. Interleukin-22 Ameliorates Neutrophil-Driven Nonalcoholic Steatohepatitis Through Multiple Targets. Hepatology 2020, 72, 412–429. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neuckermans, J.; Lequeue, S.; Claes, P.; Heymans, A.; Hughes, J.H.; Colemonts-Vroninks, H.; Marcélis, L.; Casimir, G.; Goyens, P.; Martens, G.A.; et al. Hereditary Tyrosinemia Type 1 Mice under Continuous Nitisinone Treatment Display Remnants of an Uncorrected Liver Disease Phenotype. Genes 2023, 14, 693. https://doi.org/10.3390/genes14030693
Neuckermans J, Lequeue S, Claes P, Heymans A, Hughes JH, Colemonts-Vroninks H, Marcélis L, Casimir G, Goyens P, Martens GA, et al. Hereditary Tyrosinemia Type 1 Mice under Continuous Nitisinone Treatment Display Remnants of an Uncorrected Liver Disease Phenotype. Genes. 2023; 14(3):693. https://doi.org/10.3390/genes14030693
Chicago/Turabian StyleNeuckermans, Jessie, Sien Lequeue, Paul Claes, Anja Heymans, Juliette H. Hughes, Haaike Colemonts-Vroninks, Lionel Marcélis, Georges Casimir, Philippe Goyens, Geert A. Martens, and et al. 2023. "Hereditary Tyrosinemia Type 1 Mice under Continuous Nitisinone Treatment Display Remnants of an Uncorrected Liver Disease Phenotype" Genes 14, no. 3: 693. https://doi.org/10.3390/genes14030693
APA StyleNeuckermans, J., Lequeue, S., Claes, P., Heymans, A., Hughes, J. H., Colemonts-Vroninks, H., Marcélis, L., Casimir, G., Goyens, P., Martens, G. A., Gallagher, J. A., Vanhaecke, T., Bou-Gharios, G., & De Kock, J. (2023). Hereditary Tyrosinemia Type 1 Mice under Continuous Nitisinone Treatment Display Remnants of an Uncorrected Liver Disease Phenotype. Genes, 14(3), 693. https://doi.org/10.3390/genes14030693