
High Throughput SARS-CoV-2 Genome Sequencing from 384 Respiratory Samples Using the Illumina COVIDSeq Protocol

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Samples
2.2. RNA Extraction
2.3. Standard Illumina CovidSeq Protocol
2.4. Modified Illumina COVIDSeq Protocol
2.5. NGS Data Analysis
2.5.1. Standard Illumina COVIDSeq Sequencing Data
2.5.2. Modified Illumina MiSeq Sequencing Data
2.6. Statistical Analysis
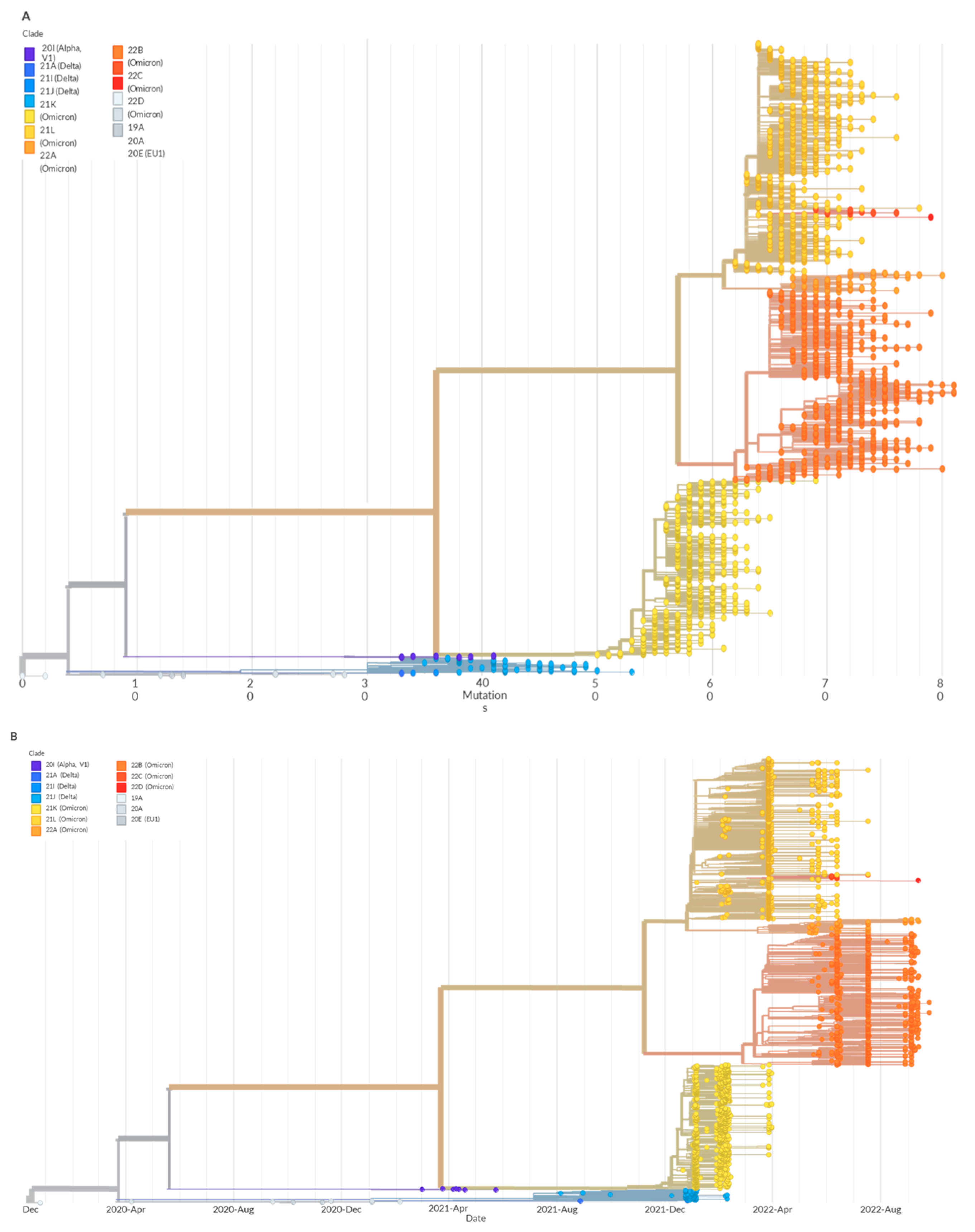
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- World Health Organization. Listings of WHO’s Response to COVID-19. Available online: https://www.who.int/news/item/29-06-2020-covidtimeline (accessed on 2 March 2023).
- Stoecklin, S.B.; Rolland, P.; Silue, Y.; Mailles, A.; Campese, C.; Simondon, A.; Mechain, M.; Meurice, L.; Nguyen, M.; Bassi, C.; et al. First cases of coronavirus disease 2019 (COVID-19) in France: Surveillance, investigations and control measures, January 2020. Euro Surveill. 2020, 25, 2000094. [Google Scholar]
- World Health Organization. France: WHO Coronavirus Disease (COVID-19) Dashboard with Vaccination Data. Available online: https://covid19.who.int/region/euro/country/fr (accessed on 13 February 2023).
- Santé Publique France. InfoCovidFrance: Chiffres clés et Evolution de la COVID-19 en France et dans le Monde. Available online: https://www.santepubliquefrance.fr/dossiers/coronavirus-covid-19/coronavirus-chiffres-cles-et-evolution-de-la-covid-19-en-france-et-dans-le-monde (accessed on 13 February 2023).
- Bhoyar, R.C.; Senthivel, V.; Jolly, B.; Imran, M.; Jain, A.; Divakar, M.K.; Scaria, V.; Sivasubbu, S. An Optimized, Amplicon-Based Approach for Sequencing of SARS-CoV-2 from Patient Samples using COVIDSeq Assay on ILLUMINA MiSeq Sequencing Platforms. STAR Protoc. 2021, 2, 100755. [Google Scholar] [CrossRef] [PubMed]
- Papa Mze, N.; Beye, M.; Kacel, I.; Tola, R.; Basco, L.; Bogreau, H.; Colson, P.; Fournier, P.E. Simultaneous SARS-CoV-2 genome sequencing of 384 samples on an Illumina MiSeq instrument through protocol optimization. Genes 2022, 13, 1648. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Radulescu, C.; Tang, M.; Mahesh, A.; Lavin, D.; Umbreen, S.; McKenna, J.; Smyth, M.; McColgan, E.; Molnar, Z.; et al. Mini-XT, a miniaturized tagmentation-based protocol for efficient sequencing of SARS-CoV-2. J. Transl. Med. 2022, 20, 105. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.E.; Colson, P.; Levasseur, A.; Devaux, C.A.; Gautret, P.; Bedotto, M.; Delerce, J.; Brechard, L.; Pinaul, L.; Lagier, J.C.; et al. Emergence and outcomes of the SARS-CoV-2 ‘Marseille-4’ variant. Int. J. Infect. Dis. 2021, 106, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 12 October 2022).
- DeGrace, M.M.; Ghedin, E.; Frieman, M.B.; Krammer, F.; Grifoni, A.; Alisoltani, A.; Alter, G.; Amara, R.R.; Baric, R.S.; Barouch, D.H.; et al. Defining the risk of SARS-CoV-2 variants on immune protection. Nature 2022, 605, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Santé Publique France. Consortium EMERGEN—Activité Hebdomadaire de Séquençage. Available online: www.santepubliquefrance.fr/dossiers/coronavirus-covid-19/coronavirus-circulation-des-variants-du-sars-cov-2#block-338801 (accessed on 11 September 2022).
- Hodcroft, E. CoVariants: SARS-CoV-2 Mutations and Variants of Interest. 2021. Available online: https://covariants.org/ (accessed on 11 September 2022).
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brunink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. EuroSurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed]
- Santé Publique France. Consortium EMERGEN. Available online: https://www.santepubliquefrance.fr/dossiers/coronavirus-covid-19/consortium-emergen (accessed on 27 September 2022).
- ARTIC Network. Real-Time Molecular Epidemiology for Outbreak Response. Available online: https://artic.network (accessed on 11 September 2022).
- Colson, P.; Fournier, P.E.; Chaudet, H.; Delerce, J.; Giraud-Gatineau, A.; Houhamdi, L.; Andrieu, C.; Brechard, L.; Bedotto, M.; Prudent, E. Analysis of SARS-CoV-2 variants from 24,181 patients exemplifies the role of globalization and zoonosis in pandemics. Front. Microbiol. 2022, 12, 786233. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; Fournier, P.E.; Delerce, J.; Million, M.; Bedotto, M.; Houhamdi, L.; Yahi, N.; Bayette, J.; Levasseur, A.; Fantini, J.; et al. Culture and identification of a “Deltamicron” SARS-CoV-2 in a three cases cluster in southern France. J. Med. Virol. 2022, 94, 3739–3749. [Google Scholar] [CrossRef] [PubMed]
- GitHub. v2.2.1. Available online: https://github.com/bwa-mem2/bwa-mem2 (accessed on 12 September 2022).
- SAMtools. Available online: https://www.htslib.org (accessed on 12 September 2022).
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- GitHub. Available online: https://github.com/freebayes/freebayes (accessed on 12 September 2022).
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-ead sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- BCFtools. Available online: https://samtools.github.io/bcftools/bcftools.html (accessed on 12 September 2022).
- Nextclade Web 2.12.0, Nextclade CLI 2.12.0. Available online: https://clades.nextstrain.org (accessed on 12 September 2022).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN). Available online: https://cov-lineages.org/pangolin.html (accessed on 12 September 2022).
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2022, 50, D161–D164. [Google Scholar] [CrossRef] [PubMed]
- Global Initiative on Sharing Avian Influenza Data (GISAID). Available online: https://gisaid.org/ (accessed on 12 September 2022).


{kind=link}
| Methods | Number of Amplicons Tested | Volume of Library (µL) | Cluster Density (K/mm²) | Passing Filter (%) | Estimated Yield (Mb) | Q30 (G/%) |
|---|---|---|---|---|---|---|
| Bhoyar et al. 2021 [5] | 96 | 5 | unknown | unknown | unknown | unknown |
| COVIDSeq modified protocol | 96 | 5 | 1005 | 88.3 | 8862 | 6.9/81.6 |
| 192 | 7 | 1013 | 92.0 | 9274 | 7.2/81.5 | |
| 384 | 7 | 1347 | 81.4 | 10,244 | 7.5/76.5 |
| Technique | 20A | 20I | 21I | 21J | 21K | 21L | Total |
|---|---|---|---|---|---|---|---|
| MiSeq | 2 | 5 | 4 | 65 | 279 | 1 | 356 |
| NovaSeq | 2 | 5 | 4 | 66 | 279 | 1 | 357 |
| Technique | Reagent | Manufacturer | Total Duration (h) | Cost (euros) | Total Cost (euros) | |
|---|---|---|---|---|---|---|
| NovaSeq | MiSeq | |||||
| protocol modified for COVIDSeq and MiSeq (for preparation of library) | Illumina COVIDSeq™ Test (384 Samples) | Illumina | 8 | 4212 | 8025 | 6794 |
| IDT for Illumina PCR Indexes Set 1–4 | Illumina | 1267 | ||||
| Illumina COVIDSeq v4 Primer Pools, 384 Samples RUO | Illumina | 235 | ||||
| Qubit dsDNA HS Assay Kit | Thermo Fisher | 60 | ||||
| NovaSeq | NovaSeq 6000 SP Reagent Kit (100 cycles) V1.5 | Illumina | 14 | 1971 | ||
| NovaSeq XP 2-Lane Kit V1.5 | Illumina | 280 | ||||
| MiSeq | MiSeq Reagent Kit V2 (500 cycles) | Illumina | 44 | 1020 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papa Mze, N.; Kacel, I.; Beye, M.; Tola, R.; Sarr, M.; Basco, L.; Bogreau, H.; Colson, P.; Fournier, P.-E. High Throughput SARS-CoV-2 Genome Sequencing from 384 Respiratory Samples Using the Illumina COVIDSeq Protocol. Genes 2023, 14, 681. https://doi.org/10.3390/genes14030681
Papa Mze N, Kacel I, Beye M, Tola R, Sarr M, Basco L, Bogreau H, Colson P, Fournier P-E. High Throughput SARS-CoV-2 Genome Sequencing from 384 Respiratory Samples Using the Illumina COVIDSeq Protocol. Genes. 2023; 14(3):681. https://doi.org/10.3390/genes14030681
Chicago/Turabian StylePapa Mze, Nasserdine, Idir Kacel, Mamadou Beye, Raphael Tola, Mariéma Sarr, Leonardo Basco, Hervé Bogreau, Philippe Colson, and Pierre-Edouard Fournier. 2023. "High Throughput SARS-CoV-2 Genome Sequencing from 384 Respiratory Samples Using the Illumina COVIDSeq Protocol" Genes 14, no. 3: 681. https://doi.org/10.3390/genes14030681
APA StylePapa Mze, N., Kacel, I., Beye, M., Tola, R., Sarr, M., Basco, L., Bogreau, H., Colson, P., & Fournier, P.-E. (2023). High Throughput SARS-CoV-2 Genome Sequencing from 384 Respiratory Samples Using the Illumina COVIDSeq Protocol. Genes, 14(3), 681. https://doi.org/10.3390/genes14030681