
MicroRNA Target Identification: Revisiting Accessibility and Seed Anchoring

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.1.1. Human CLASH Data
2.1.2. Mouse CLASH Data
2.2. Revisiting Accessibility through CLASH Data
- The seed match. We recall that this is the problem of finding the best binding site between two RNAs. It is addressed by adopting a sequence alignment method, albeit with some adaptation, the main of which is that sequence complementarity and not sequence identity is considered. We recall that in the case of miRNAs, the seed starts at nucleotide 2 from the 5 end of the miRNA and is of a length between 6 and 8.
- The minimum free energy of the interaction, or what was called the stability of the duplex formed between the miRNA and the mRNA in the introduction. This corresponds to the secondary structure between miRNA and target mRNA that has the lowest value of free energy required to unfold it. Some methods further work within a nearest-neighbour free energy model, where dinucleotides instead of single nucleotides are considered [19]. The way free energy is computed may differ importantly between different methods [20,21].
- Pita: Among the methods for predicting the targets of a miRNA, Pita was a pioneer in considering accessibility when attempting to carry out such predictions. With that aim in sight, after identifying miRNA interaction sites using complementary sequences to the seed, it computes an energy score , which is equal to the difference between duplex and open, the first being the energy for binding both RNAs together, which is computed with a modified version of RNAduplex, while the second is the energy required to open a region and is computed using RNAfold [23] and a sliding window of 70 additional nucleotides before and after the predicted target region.
- IntaRNA: Similar to Pita, IntaRNA predicts RNA–RNA interactions using the accessibility of the target sites. IntaRNA actually combines two energy scores, the first one representing the target accessibility computed with RNAplfold [23], and the second one corresponding to the energy of the RNA–RNA hybridisation computed as in RNAhybrid [20]. Again, as with Pita, IntaRNA does look for complementary sequences to the seed; however, contrarily to Pita, it does this in a third step only.
- miRanda: miRanda is the only method among the three chosen that does not consider the accessibility of the target region. The method proceeds by first searching for complementary sequences with an emphasis on binding the 5’ region of the miRNA (region of the seed) and then by computing the energy of the pairing using RNAduplex.
2.3. Performance Evaluation
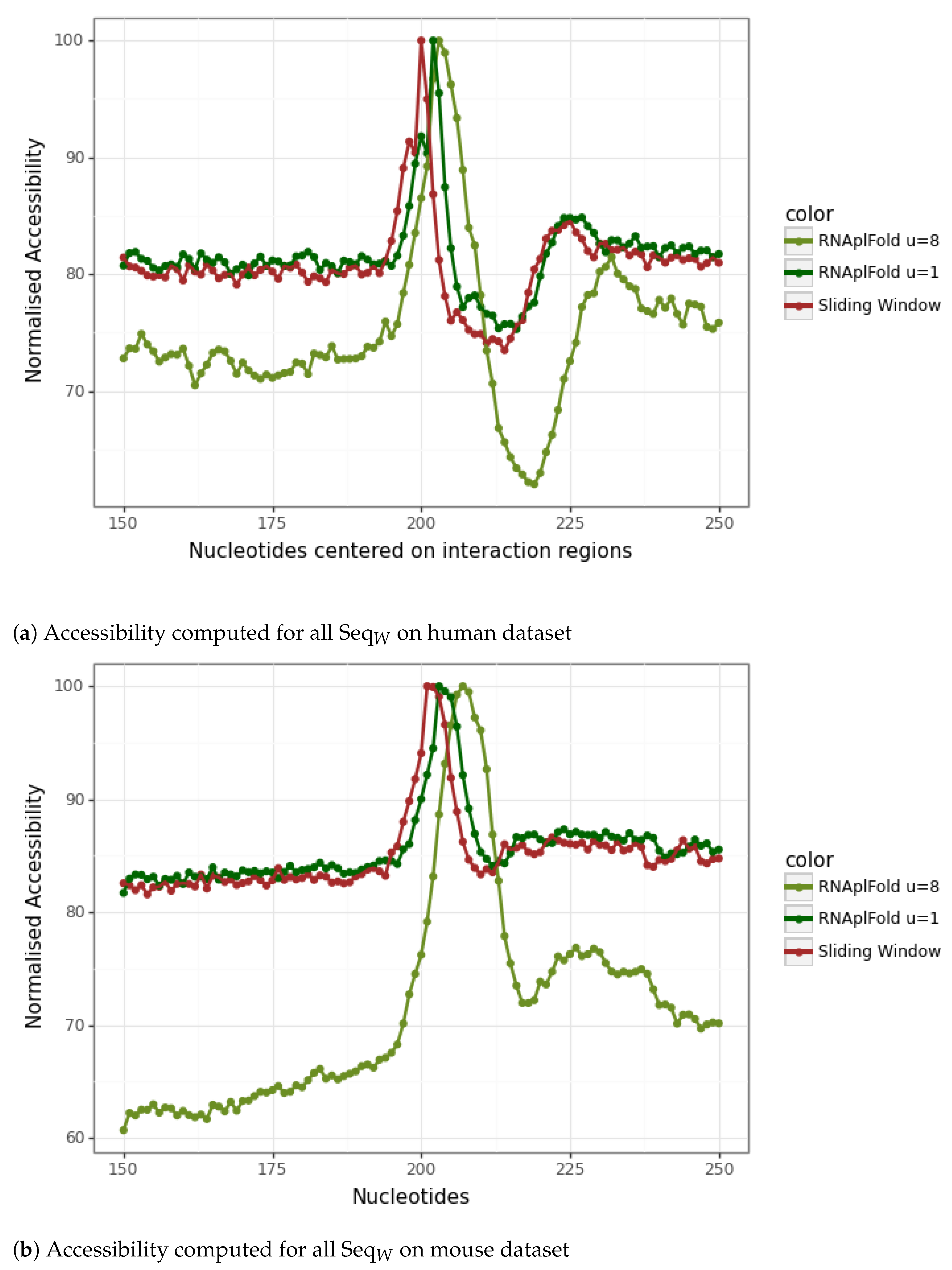
2.4. mRNA Accessibility
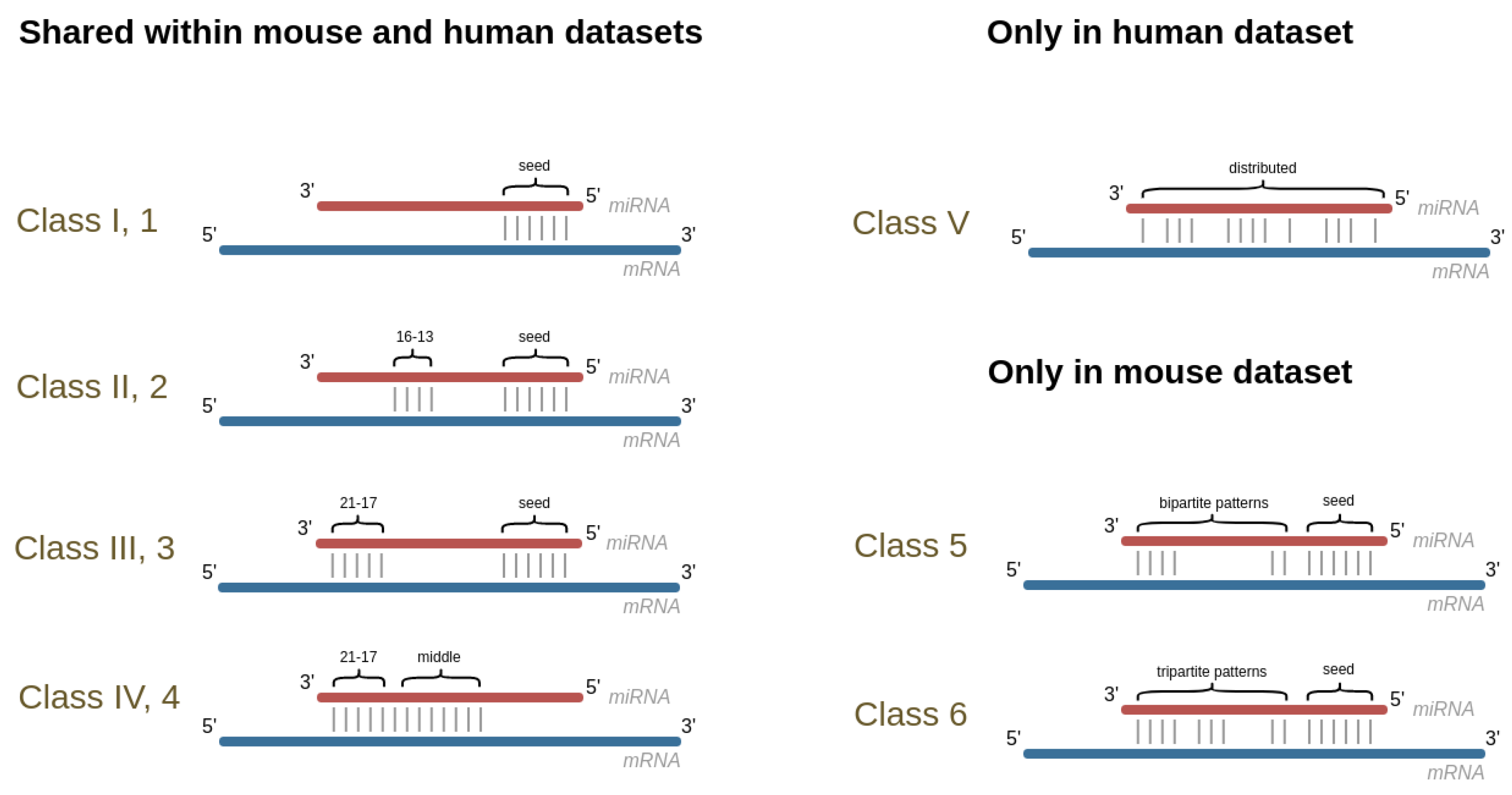
2.5. Exploring Intra-Species Conservation
3. Results
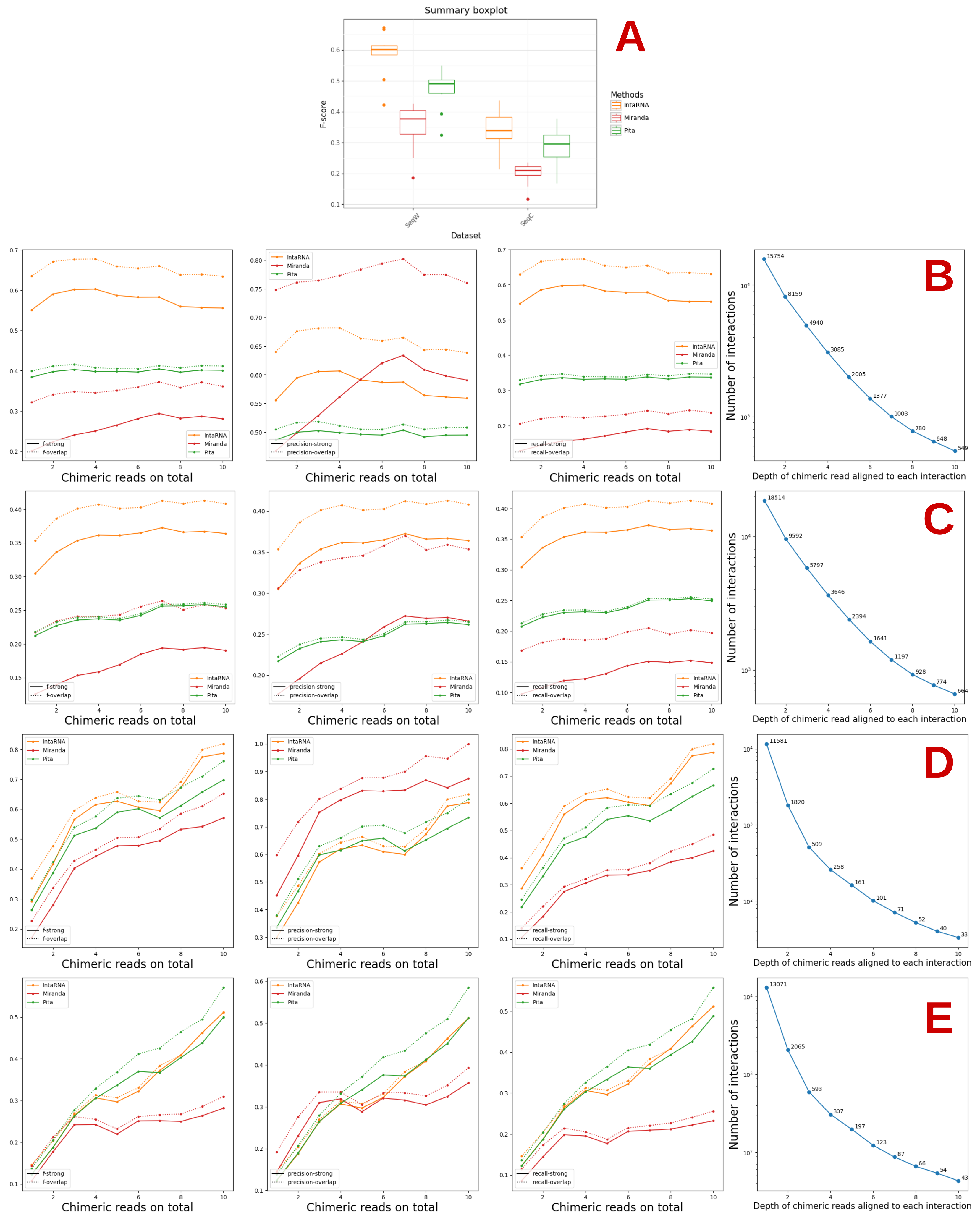
3.1. Prediction of Interactions from the CLASH Datasets Using Three Methods
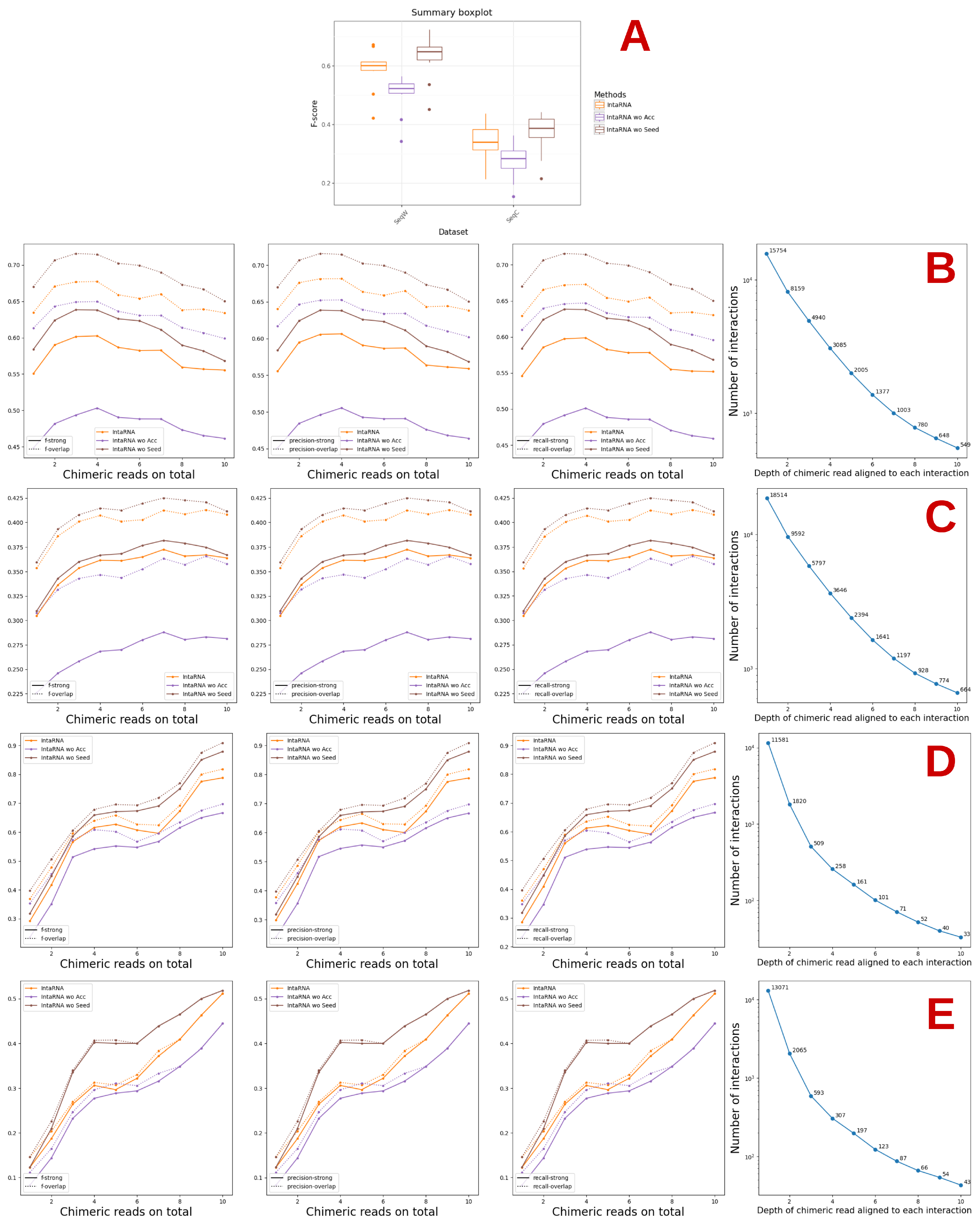
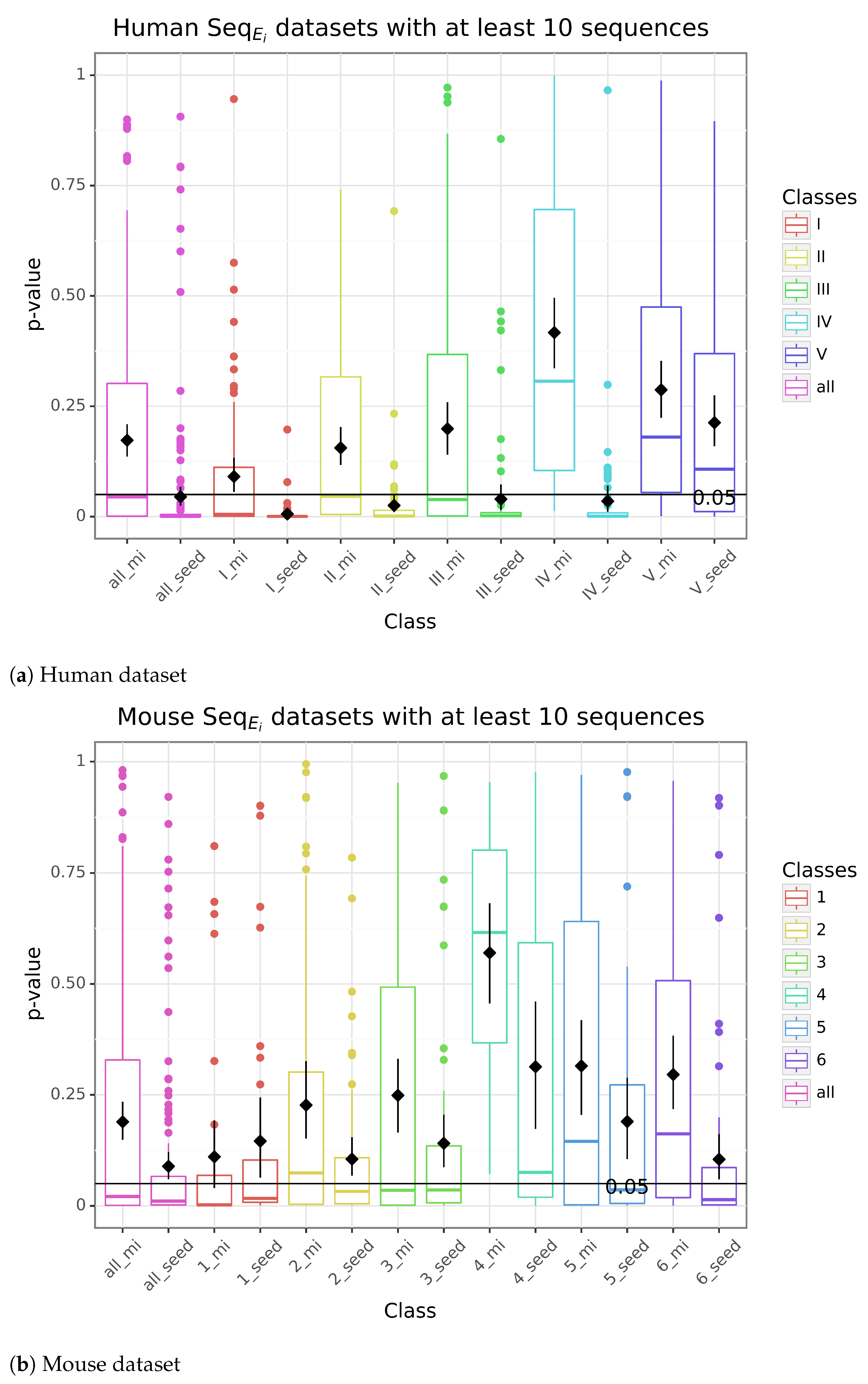
3.2. Study of the Accessibility of the Interaction Site and Seed Match with IntaRNA
3.3. Refinement of the Accessibility Study
3.4. Exploring Intra-Species Conservation
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vasudevan, S. Posttranscriptional Upregulation by MicroRNAs: Posttranscriptional Upregulation by MicroRNAs. Wiley Interdiscip. Rev. RNA 2012, 3, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. Secreted microRNAs: A new form of intercellular communication. Cell Press 2012, 22, 8. [Google Scholar] [CrossRef] [PubMed]
- Hoy, A.; Buck, A. Extracellular small RNAs: What, where, why? Biochem. Soc. Trans. 2012, 40, 886–890. [Google Scholar] [CrossRef]
- Taylor, D.D.; Gercel-Taylor, C. The origin, function, and diagnostic potential of RNA within extracellular vesicles present in human biological fluids. Front. Genet. 2013, 4, 00142. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; He, B.; Kogel, K.H.; Jin, H. Cross-kingdom RNA trafficking and environmental RNAi —Nature’s blueprint for modern crop protection strategies. Curr. Opin. Microbiol. 2018, 46, 58–64. [Google Scholar] [CrossRef]
- Liu, S.; da Cunha, A.; Rezende, R.; Cialic, R.; Wei, Z.; Bry, L.; Comstock, L.; Gandhi, R.; Weiner, H. The Host Shapes the Gut Microbiota via Fecal MicroRNA. Cell Host Microbe 2016, 19, 32–43. [Google Scholar] [CrossRef]
- Fridrich, A.; Hazan, Y.; Moran, Y. Too Many False Targets for MicroRNAs: Challenges and Pitfalls in Prediction of miRNA Targets and Their Gene Ontology in Model and Non-model Organisms. BioEssays 2019, 41, 1800169. [Google Scholar] [CrossRef]
- Mockly, S.; Seitz, H. Inconsistencies and Limitations of Current MicroRNA Target Identification Methods. In MicroRNA Target Identification; Lagana, A., Ed.; Springer: New York, NY, USA, 2019; Volume 1970, pp. 291–314. [Google Scholar] [CrossRef]
- Hutvagner, G.; Simard, M.J.; Mello, C.C.; Zamore, P.D. Sequence-Specific Inhibition of Small RNA Function. PLoS Biol. 2004, 2, e98. [Google Scholar] [CrossRef]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the Human miRNA Interactome by CLASH Reveals Frequent Noncanonical Binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef]
- Moore, M.J.; Scheel, T.K.H.; Luna, J.M.; Park, C.Y.; Fak, J.J.; Nishiuchi, E.; Rice, C.M.; Darnell, R.B. miRNA-target chimeras reveal miRNA 3′-end pairing as a major determinant of Argonaute target specificity. Nat. Commun. 2015, 6, 8864. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, R.J.; Kahari, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef] [PubMed]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA Targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Richter, A.S.; Backofen, R. IntaRNA: Efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 2008, 24, 2849–2856. [Google Scholar] [CrossRef]
- Mann, M.; Wright, P.R.; Backofen, R. IntaRNA 2.0: Enhanced and customizable prediction of RNA-RNA interactions. Nucleic Acids Res. 2017, 45, W435–W439. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Honer zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Turner, D.H.; Mathews, D.H. NNDB: The nearest neighbor parameter database for predicting stability of nucleic acid secondary structure. Nucleic Acids Res. 2010, 38, D280–D282. [Google Scholar] [CrossRef]
- Kruger, J.; Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef]
- Tafer, H.; Hofacker, I.L. RNAplex: A fast tool for RNA-RNA interaction search. Bioinformatics 2008, 24, 2657–2663. [Google Scholar] [CrossRef]
- Marin, R.M.; Vanicek, J. Efficient use of accessibility in microRNA target prediction. Nucleic Acids Res. 2011, 39, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, L.S.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Monatshefte Chem. Chem. Mon. 1994, 125, 167–188. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Gribskov, M. Combining evidence using p-values: Application to sequence homology searches. Bioinformatics 1998, 14, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.S.; Tam, W.L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A Pattern-Based Method for the Identification of MicroRNA Binding Sites and Their Corresponding Heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of interactions for the Seq dataset | |||||||||
| Total | 5UTR | CDS | 3UTR | Other mRNA regions | Class I | Class II | Class III | Class IV | Class V |
| 18,497 | 868 | 11,112 | 6093 | 424 | 3590 | 3290 | 4629 | 3382 | 3606 |
| Number of interactions for the Seq dataset | |||||||||
| Total | 5UTR | CDS | 3UTR | Other mRNA regions | Class I | Class II | Class III | Class IV | Class V |
| 15,754 | 291 | 10,304 | 4916 | 243 | 3095 | 2833 | 3970 | 2890 | 2966 |
| Number of interactions for the Seq dataset | ||||||||||
| Total | 3UTR | CDS | 5UTR | Other mRNA regions | Class 1 | Class 2 | Class 3 | Class 4 | Class 5 | Class 6 |
| 13,071 | 4843 | 7136 | 187 | 905 | 1631 | 2672 | 2269 | 1117 | 2363 | 3019 |
| Number of interactions for the Seq dataset | ||||||||||
| Total | 3UTR | CDS | 5UTR | Other mRNA regions | Class 1 | Class 2 | Class 3 | Class 4 | Class 5 | Class 6 |
| 11,581 | 3817 | 6917 | 102 | 745 | 1428 | 2366 | 1987 | 976 | 2129 | 2695 |
| Human Dataset | ||||
| Seq | Seq | |||
| Classes | Av. width | #Seqs | Av. width | #Seqs |
| all | 11.31 | 18,431 | 14.57 | 15,670 |
| I | 10.36 | 3531 | 13.77 | 3030 |
| II | 11.50 | 3224 | 14.83 | 2770 |
| III | 11.84 | 4560 | 15.11 | 3907 |
| IV | 11.0 | 3318 | 14.29 | 2828 |
| V | 10.15 | 3534 | 14.25 | 2898 |
| Mouse Dataset | ||||
| Seq | Seq | |||
| Classes | Av. width | #Seqs | Av. width | #Seqs |
| all | 10.0 | 12,992 | 14.72 | 11,500 |
| 1 | 9.45 | 1556 | 13.18 | 1357 |
| 2 | 9.77 | 2588 | 13.40 | 2287 |
| 3 | 9.54 | 2182 | 14.01 | 1908 |
| 4 | 8.92 | 1061 | 14.35 | 922 |
| 5 | 9.90 | 2298 | 13.62 | 2057 |
| 6 | 9.42 | 2947 | 14.09 | 2624 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Homberg, N.; Galvão Ferrarini, M.; Gaspin, C.; Sagot, M.-F. MicroRNA Target Identification: Revisiting Accessibility and Seed Anchoring. Genes 2023, 14, 664. https://doi.org/10.3390/genes14030664
Homberg N, Galvão Ferrarini M, Gaspin C, Sagot M-F. MicroRNA Target Identification: Revisiting Accessibility and Seed Anchoring. Genes. 2023; 14(3):664. https://doi.org/10.3390/genes14030664
Chicago/Turabian StyleHomberg, Nicolas, Mariana Galvão Ferrarini, Christine Gaspin, and Marie-France Sagot. 2023. "MicroRNA Target Identification: Revisiting Accessibility and Seed Anchoring" Genes 14, no. 3: 664. https://doi.org/10.3390/genes14030664
APA StyleHomberg, N., Galvão Ferrarini, M., Gaspin, C., & Sagot, M.-F. (2023). MicroRNA Target Identification: Revisiting Accessibility and Seed Anchoring. Genes, 14(3), 664. https://doi.org/10.3390/genes14030664