
Epigenetic Regulation of β-Globin Genes and the Potential to Treat Hemoglobinopathies through Epigenome Editing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
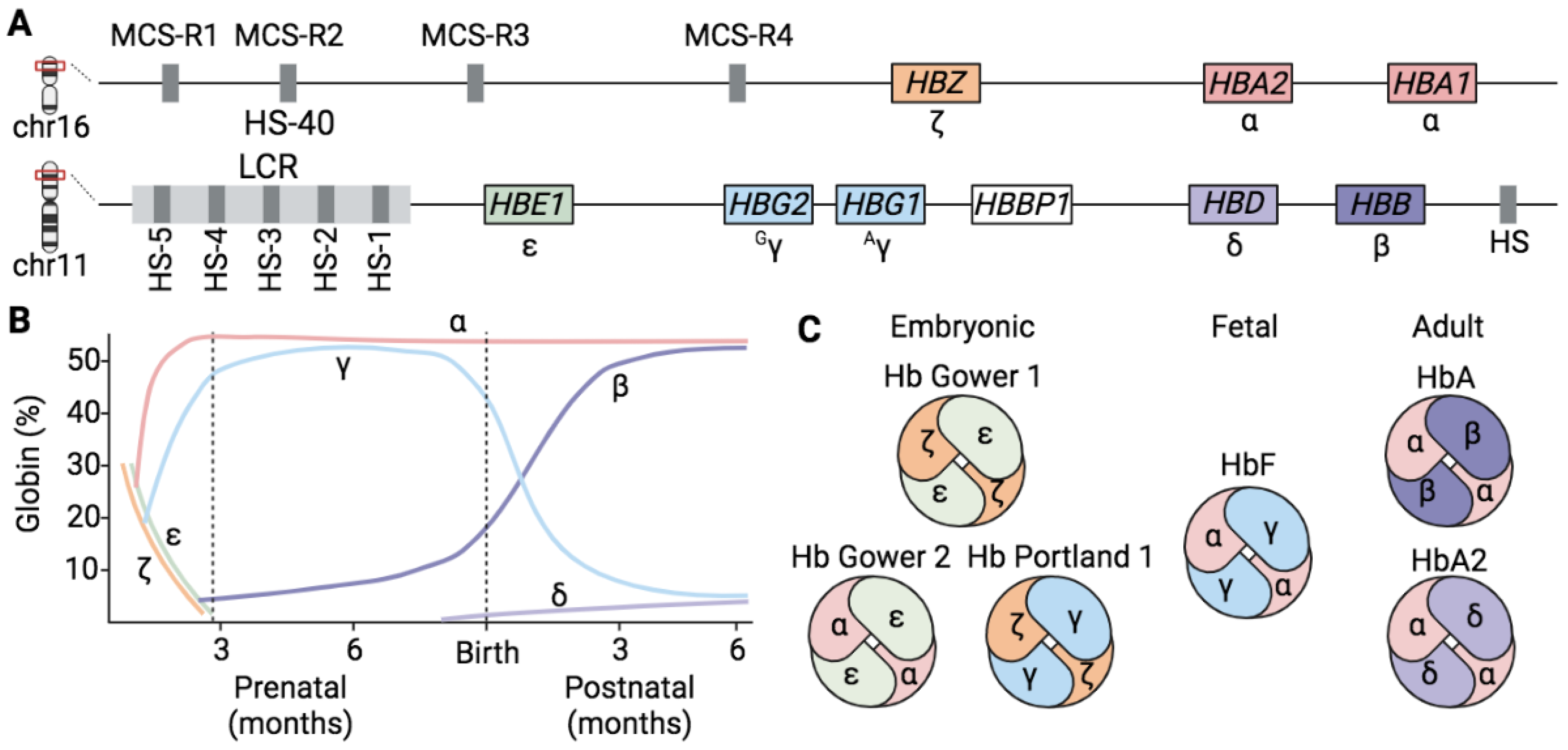
1.1. Hemoglobins
1.2. Globin Gene Regulation
1.3. The Fetal-to-Adult Hb Switching
1.4. Beta-Hemoglobinopathies
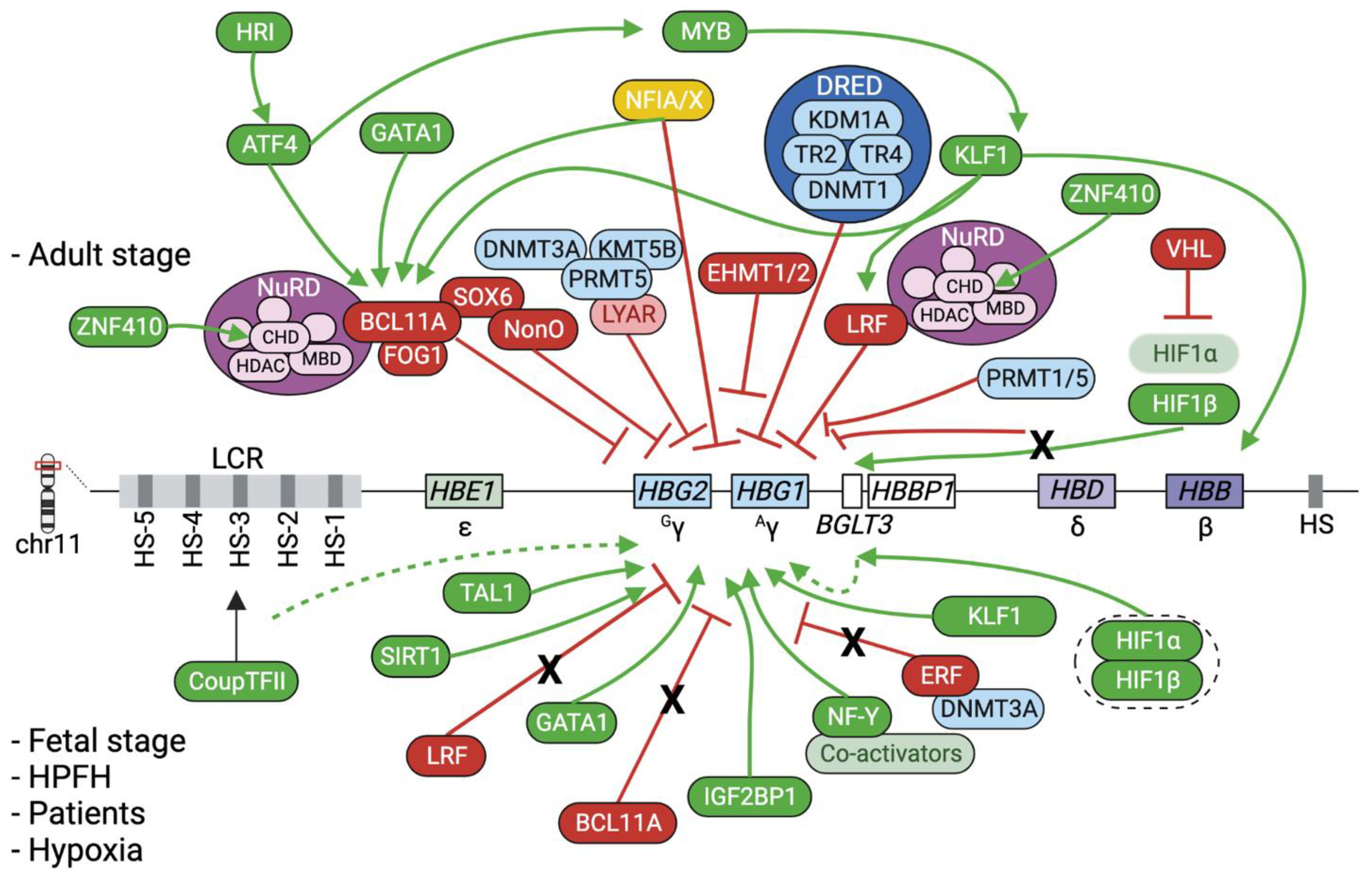
2. Epigenetic Regulation of the β-Globin Genes
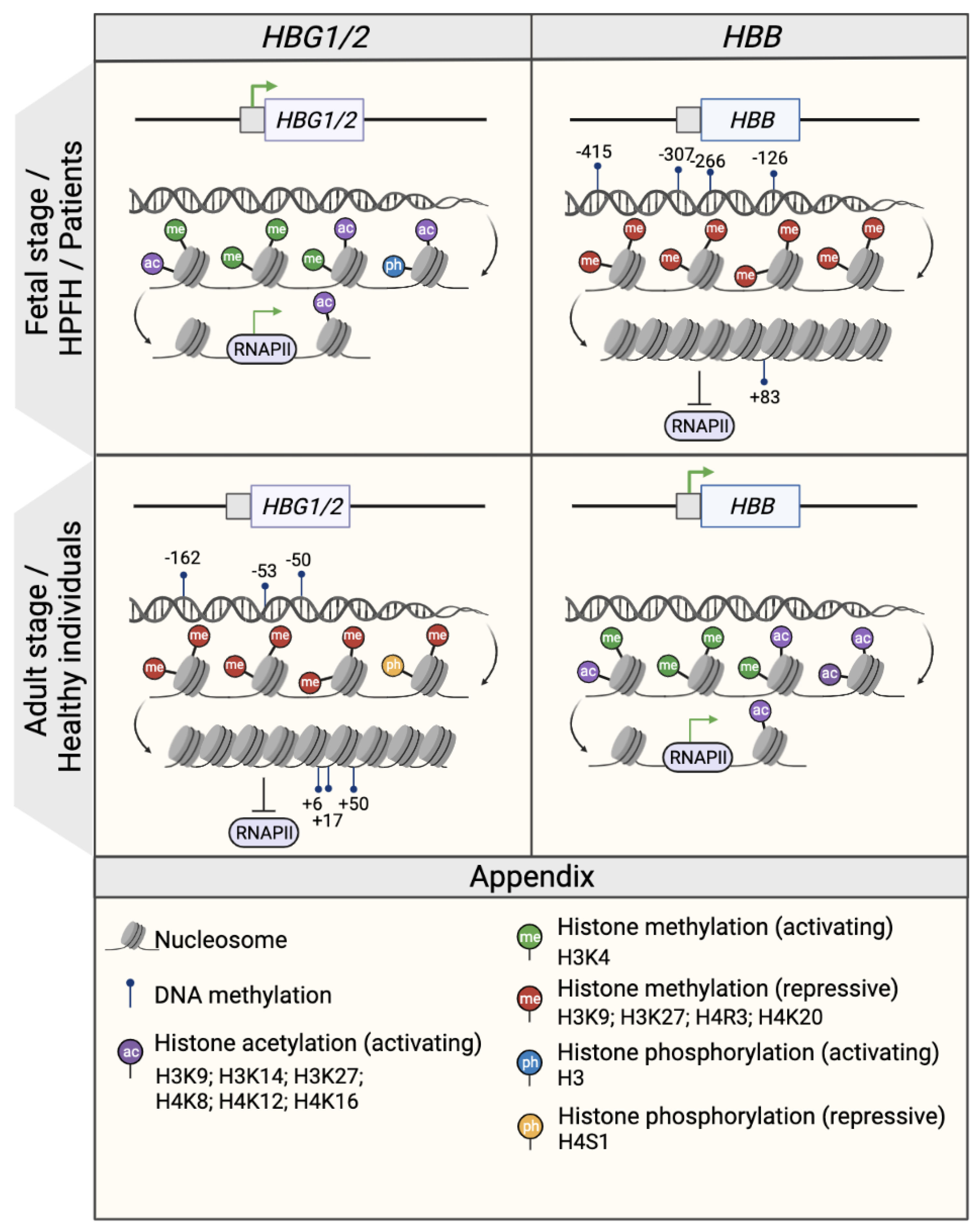
2.1. DNA Methylation
2.2. Histone Modifications
2.2.1. Histone Methylations
2.2.2. Histone Acetylations
2.3. The Interplay between Epigenetic Modifiers, Transcription Factors and Chromatin Looping
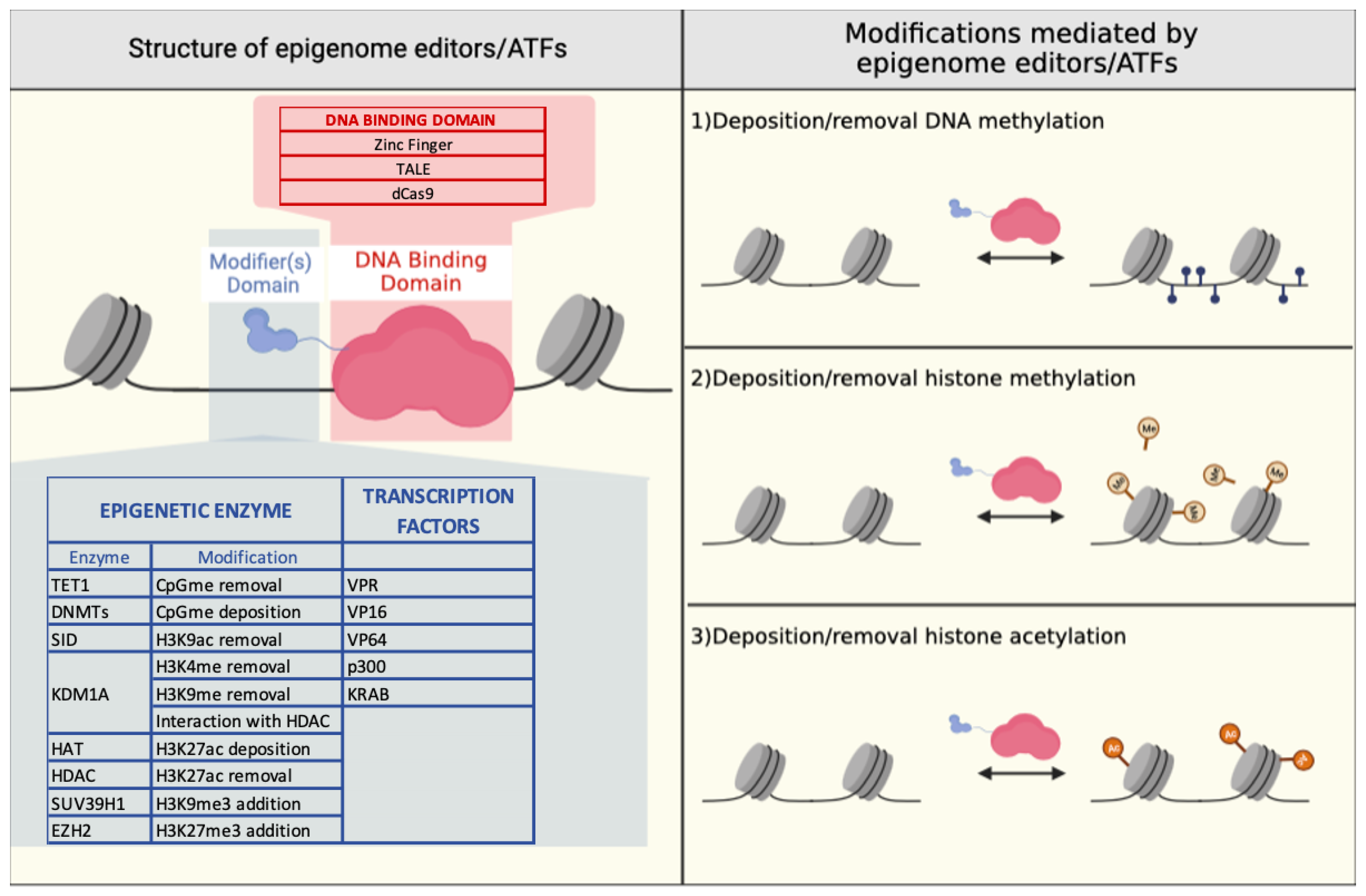
3. Epigenome Editing
3.1. Zinc Finger-Based Epigenome Editors
3.2. TALE-Based Epigenome Editors
3.3. CRISPR/Cas9-Based Epigenome Editors
3.4. Epigenetic Approaches to Modulate β-like Globin Expression
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Manning, L.R.; Russell, J.E.; Padovan, J.C.; Chait, B.T.; Popowicz, A.; Manning, R.S.; Manning, J.M. Human Embryonic, Fetal, and Adult Hemoglobins Have Different Subunit Interface Strengths. Correlation with Lifespan in the Red Cell. Protein Sci. 2007, 16, 1641–1658. [Google Scholar] [CrossRef]
- Jonxis, J.H.P. The Development of Hemoglobin. Pediatr. Clin. N. Am. 1965, 12, 535–550. [Google Scholar] [CrossRef]
- Lorkin, P.A. Fetal and Embryonic Haemoglobins. J. Med. Genet. 1973, 10, 50–64. [Google Scholar] [CrossRef]
- Maston, G.A.; Evans, S.K.; Green, M.R. Transcriptional Regulatory Elements in the Human Genome. Annu. Rev. Genom. Hum. Genet. 2006, 7, 29–59. [Google Scholar] [CrossRef] [PubMed]
- Perutz, M.F. Mechanisms of Cooperativity and Allosteric Regulation in Proteins. Quart. Rev. Biophys. 1989, 22, 139–237. [Google Scholar] [CrossRef]
- Manning, L.R.; Russell, J.E.; Popowicz, A.M.; Manning, R.S.; Padovan, J.C.; Manning, J.M. Energetic Differences at the Subunit Interfaces of Normal Human Hemoglobins Correlate with Their Developmental Profile. Biochemistry 2009, 48, 7568–7574. [Google Scholar] [CrossRef] [PubMed]
- Efstratiadis, A. The Structure and Evolution of the Human β-Globin Gene Family. Cell 1980, 21, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Mettananda, S.; Gibbons, R.J.; Higgs, D.R. Understanding α-Globin Gene Regulation and Implications for the Treatment of β-Thalassemia: α-Globin Regulation and β-Thalassemia. Ann. N. Y. Acad. Sci. 2016, 1368, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Martyn, G.E.; Quinlan, K.G.R.; Crossley, M. The Regulation of Human Globin Promoters by CCAAT Box Elements and the Recruitment of NF-Y. Biochim. Et Biophys. Acta (BBA) Gene Regul. Mech. 2017, 1860, 525–536. [Google Scholar] [CrossRef]
- Philipsen, S.; Talbot, D.; Fraser, P.; Grosveld, F. The Beta-Globin Dominant Control Region: Hypersensitive Site 2. EMBO J. 1990, 9, 2159–2167. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Chen, Y.; Li, M.; Zhou, F.; Li, K.; Cao, H.; Ni, M.; Liu, Y.; Gu, Z.; et al. In Situ Capture of Chromatin Interactions by Biotinylated DCas9. Cell 2017, 170, 1028–1043.e19. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Stamatoyannopoulos, G. Hypersensitive Site 5 of the Human Beta Locus Control Region Functions as a Chromatin Insulator. Blood 1994, 84, 1399–1401. [Google Scholar] [CrossRef]
- Himadewi, P.; Wang, X.Q.D.; Feng, F.; Gore, H.; Liu, Y.; Yu, L.; Kurita, R.; Nakamura, Y.; Pfeifer, G.P.; Liu, J.; et al. 3’HS1 CTCF Binding Site in Human β-Globin Locus Regulates Fetal Hemoglobin Expression. Elife 2021, 10, e70557. [Google Scholar] [CrossRef] [PubMed]
- Talbot, D.; Philipsen, S.; Fraser, P.; Grosveld, F. Detailed Analysis of the Site 3 Region of the Human Beta-Globin Dominant Control Region. EMBO J. 1990, 9, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Qin, K.; Huang, P.; Feng, R.; Keller, C.A.; Peslak, S.A.; Khandros, E.; Saari, M.S.; Lan, X.; Mayuranathan, T.; Doerfler, P.A.; et al. Dual Function NFI Factors Control Fetal Hemoglobin Silencing in Adult Erythroid Cells. Nat. Genet. 2022, 54, 874–884. [Google Scholar] [CrossRef]
- Bao, X.; Zhang, X.; Wang, L.; Wang, Z.; Huang, J.; Zhang, Q.; Ye, Y.; Liu, Y.; Chen, D.; Zuo, Y.; et al. Epigenetic Inactivation of ERF Reactivates γ-Globin Expression in β-Thalassemia. Am. J. Hum. Genet. 2021, 108, 709–721. [Google Scholar] [CrossRef]
- Fugazza, C.; Barbarani, G.; Elangovan, S.; Marini, M.G.; Giolitto, S.; Font-Monclus, I.; Marongiu, M.F.; Manunza, L.; Strouboulis, J.; Cantù, C.; et al. The Coup-TFII Orphan Nuclear Receptor Is an Activator of the γ-Globin Gene. Haematologica 2020, 106, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.B.; Gross, J.; Pratt, K.; Guo, X.; Byrnes, C.; Lee, Y.T.; Lavelle, D.; Dean, A.; Miller, J.L.; Wilber, A. The MRNA-Binding Protein IGF2BP1 Restores Fetal Hemoglobin in Cultured Erythroid Cells from Patients with β-Hemoglobin Disorders. Mol. Ther. Methods Clin. Dev. 2020, 17, 429–440. [Google Scholar] [CrossRef]
- Feng, R.; Mayuranathan, T.; Huang, P.; Doerfler, P.A.; Li, Y.; Yao, Y.; Zhang, J.; Palmer, L.E.; Mayberry, K.; Christakopoulos, G.E.; et al. Activation of γ-Globin Expression by Hypoxia-Inducible Factor 1α. Nature 2022, 610, 783–790. [Google Scholar] [CrossRef]
- Doerfler, P.A.; Feng, R.; Li, Y.; Palmer, L.E.; Porter, S.N.; Bell, H.W.; Crossley, M.; Pruett-Miller, S.M.; Cheng, Y.; Weiss, M.J. Activation of γ-Globin Gene Expression by GATA1 and NF-Y in Hereditary Persistence of Fetal Hemoglobin. Nat. Genet. 2021, 53, 1177–1186. [Google Scholar] [CrossRef]
- Liu, N.; Xu, S.; Yao, Q.; Zhu, Q.; Kai, Y.; Hsu, J.Y.; Sakon, P.; Pinello, L.; Yuan, G.-C.; Bauer, D.E.; et al. Transcription Factor Competition at the γ-Globin Promoters Controls Hemoglobin Switching. Nat. Genet. 2021, 53, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Wienert, B.; Martyn, G.E.; Kurita, R.; Nakamura, Y.; Quinlan, K.G.R.; Crossley, M. KLF1 Drives the Expression of Fetal Hemoglobin in British HPFH. Blood 2017, 130, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Liu, K.; Sun, C.-W.; Pawlik, K.M.; Townes, T.M. KLF1 Regulates BCL11A Expression and γ- to β-Globin Gene Switching. Nat. Genet. 2010, 42, 742–744. [Google Scholar] [CrossRef]
- Grevet, J.D.; Lan, X.; Hamagami, N.; Edwards, C.R.; Sankaranarayanan, L.; Ji, X.; Bhardwaj, S.K.; Face, C.J.; Posocco, D.F.; Abdulmalik, O.; et al. Domain-Focused CRISPR Screen Identifies HRI as a Fetal Hemoglobin Regulator in Human Erythroid Cells. Science 2018, 361, 285–290. [Google Scholar] [CrossRef]
- Huang, P.; Peslak, S.A.; Lan, X.; Khandros, E.; Yano, J.A.; Sharma, M.; Keller, C.A.; Giardine, B.; Qin, K.; Abdulmalik, O.; et al. The HRI-Regulated Transcription Factor ATF4 Activates BCL11A Transcription to Silence Fetal Hemoglobin Expression. Blood 2020, 135, 2121–2132. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sankaran, V.G.; Ni, M.; Menne, T.F.; Puram, R.V.; Kim, W.; Orkin, S.H. Transcriptional Silencing of γ-Globin by BCL11A Involves Long-Range Interactions and Cooperation with SOX6. Genes Dev. 2010, 24, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Norton, L.J.; Funnell, A.P.W.; Burdach, J.; Wienert, B.; Kurita, R.; Nakamura, Y.; Philipsen, S.; Pearson, R.C.M.; Quinlan, K.G.R.; Crossley, M. KLF1 Directly Activates Expression of the Novel Fetal Globin Repressor ZBTB7A/LRF in Erythroid Cells. Blood Adv. 2017, 1, 685–692. [Google Scholar] [CrossRef]
- Bianchi, E.; Zini, R.; Salati, S.; Tenedini, E.; Norfo, R.; Tagliafico, E.; Manfredini, R.; Ferrari, S. C-Myb Supports Erythropoiesis through the Transactivation of KLF1 and LMO2 Expression. Blood 2010, 116, e99–e110. [Google Scholar] [CrossRef]
- Boontanrart, M.Y.; Schröder, M.S.; Stehli, G.M.; Banović, M.; Wyman, S.K.; Lew, R.J.; Bordi, M.; Gowen, B.G.; DeWitt, M.A.; Corn, J.E. ATF4 Regulates MYB to Increase γ-Globin in Response to Loss of β-Globin. Cell Rep. 2020, 32, 107993. [Google Scholar] [CrossRef]
- Lan, X.; Ren, R.; Feng, R.; Ly, L.C.; Lan, Y.; Zhang, Z.; Aboreden, N.; Qin, K.; Horton, J.R.; Grevet, J.D.; et al. ZNF410 Uniquely Activates the NuRD Component CHD4 to Silence Fetal Hemoglobin Expression. Mol. Cell 2021, 81, 239–254.e8. [Google Scholar] [CrossRef]
- Vinjamur, D.S.; Yao, Q.; Cole, M.A.; McGuckin, C.; Ren, C.; Zeng, J.; Hossain, M.; Luk, K.; Wolfe, S.A.; Pinello, L.; et al. ZNF410 Represses Fetal Globin by Singular Control of CHD4. Nat. Genet. 2021, 53, 719–728. [Google Scholar] [CrossRef]
- Wadman, I.A. The LIM-Only Protein Lmo2 Is a Bridging Molecule Assembling an Erythroid, DNA-Binding Complex Which Includes the TAL1, E47, GATA-1 and Ldb1/NLI Proteins. EMBO J. 1997, 16, 3145–3157. [Google Scholar] [CrossRef]
- Song, S.-H.; Hou, C.; Dean, A. A Positive Role for NLI/Ldb1 in Long-Range β-Globin Locus Control Region Function. Mol. Cell 2007, 28, 810–822. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Rupon, J.W.; Krivega, I.; Breda, L.; Motta, I.; Jahn, K.S.; Reik, A.; Gregory, P.D.; Rivella, S.; Dean, A.; et al. Reactivation of Developmentally Silenced Globin Genes by Forced Chromatin Looping. Cell 2014, 158, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Krivega, I.; Dale, R.K.; Dean, A. Role of LDB1 in the Transition from Chromatin Looping to Transcription Activation. Genes Dev. 2014, 28, 1278–1290. [Google Scholar] [CrossRef]
- Yun, W.J.; Kim, Y.W.; Kang, Y.; Lee, J.; Dean, A.; Kim, A. The Hematopoietic Regulator TAL1 Is Required for Chromatin Looping between the β-Globin LCR and Human γ-Globin Genes to Activate Transcription. Nucleic Acids Res. 2014, 42, 4283–4293. [Google Scholar] [CrossRef]
- Lettre, G.; Sankaran, V.G.; Bezerra, M.A.C.; Araújo, A.S.; Uda, M.; Sanna, S.; Cao, A.; Schlessinger, D.; Costa, F.F.; Hirschhorn, J.N.; et al. DNA Polymorphisms at the BCL11A, HBS1L-MYB, and β- Globin Loci Associate with Fetal Hemoglobin Levels and Pain Crises in Sickle Cell Disease. Proc. Natl. Acad. Sci. USA 2008, 105, 11869–11874. [Google Scholar] [CrossRef]
- Uda, M.; Galanello, R.; Sanna, S.; Lettre, G.; Sankaran, V.G.; Chen, W.; Usala, G.; Busonero, F.; Maschio, A.; Albai, G.; et al. Genome-Wide Association Study Shows BCL11A Associated with Persistent Fetal Hemoglobin and Amelioration of the Phenotype of β-Thalassemia. Proc. Natl. Acad. Sci. USA 2008, 105, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.E.; Kamran, S.C.; Lessard, S.; Xu, J.; Fujiwara, Y.; Lin, C.; Shao, Z.; Canver, M.C.; Smith, E.C.; Pinello, L.; et al. An Erythroid Enhancer of BCL11A Subject to Genetic Variation Determines Fetal Hemoglobin Level. Science 2013, 342, 253–257. [Google Scholar] [CrossRef]
- Liu, N.; Hargreaves, V.V.; Zhu, Q.; Kurland, J.V.; Hong, J.; Kim, W.; Sher, F.; Macias-Trevino, C.; Rogers, J.M.; Kurita, R.; et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 2018, 173, 430–442.e17. [Google Scholar] [CrossRef]
- Basak, A.; Sankaran, V.G. Regulation of the Fetal Hemoglobin Silencing Factor BCL11A: BCL11A Regulation in Hemoglobin Expression. Ann. N. Y. Acad. Sci. 2016, 1368, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Menne, T.F.; Xu, J.; Akie, T.E.; Lettre, G.; Van Handel, B.; Mikkola, H.K.A.; Hirschhorn, J.N.; Cantor, A.B.; Orkin, S.H. Human Fetal Hemoglobin Expression Is Regulated by the Developmental Stage-Specific Repressor BCL11A. Science 2008, 322, 1839–1842. [Google Scholar] [CrossRef]
- Xu, J.; Bauer, D.E.; Kerenyi, M.A.; Vo, T.D.; Hou, S.; Hsu, Y.-J.; Yao, H.; Trowbridge, J.J.; Mandel, G.; Orkin, S.H. Corepressor-Dependent Silencing of Fetal Hemoglobin Expression by BCL11A. Proc. Natl. Acad. Sci. USA 2013, 110, 6518–6523. [Google Scholar] [CrossRef] [PubMed]
- Martyn, G.E.; Wienert, B.; Yang, L.; Shah, M.; Norton, L.J.; Burdach, J.; Kurita, R.; Nakamura, Y.; Pearson, R.C.M.; Funnell, A.P.W.; et al. Natural Regulatory Mutations Elevate the Fetal Globin Gene via Disruption of BCL11A or ZBTB7A Binding. Nat. Genet. 2018, 50, 498–503. [Google Scholar] [CrossRef]
- Masuda, T.; Wang, X.; Maeda, M.; Canver, M.C.; Sher, F.; Funnell, A.P.W.; Fisher, C.; Suciu, M.; Martyn, G.E.; Norton, L.J.; et al. Transcription Factors LRF and BCL11A Independently Repress Expression of Fetal Hemoglobin. Science 2016, 351, 285–289. [Google Scholar] [CrossRef]
- Weatherall, D.J.; Clegg, J.B.; Knox-Macaulay, H.H.M.; Bunch, C.; Hopkins, C.R.; Temperley, I.J. A Genetically Determined Disorder with Features Both of Thalassaemia and Congenital Dyserythropoietic Anaemia. Br. J. Haematol. 1973, 24, 681–702. [Google Scholar] [CrossRef]
- Fernandes, Q. Therapeutic Strategies in Sickle Cell Anemia: The Past Present and Future. Life Sci. 2017, 178, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Forget, B.G. Molecular Basis of Hereditary Persistence of Fetal Hemoglobin. Ann. N. Y Acad. Sci. 1998, 850, 38–44. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted Mutation of the DNA Methyltransferase Gene Results in Embryonic Lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.A.; Rivers, A.; Ibanez, V.; Vaitkus, K.; Mahmud, N.; DeSimone, J.; Lavelle, D. Hydroxymethylcytosine and Demethylation of the γ-Globin Gene Promoter during Erythroid Differentiation. Epigenetics 2015, 10, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Charlet, J.; Duymich, C.E.; Lay, F.D.; Mundbjerg, K.; Dalsgaard Sørensen, K.; Liang, G.; Jones, P.A. Bivalent Regions of Cytosine Methylation and H3K27 Acetylation Suggest an Active Role for DNA Methylation at Enhancers. Mol. Cell 2016, 62, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Mabaera, R.; Richardson, C.A.; Johnson, K.; Hsu, M.; Fiering, S.; Lowrey, C.H. Developmental- and Differentiation-Specific Patterns of Human Gamma- and Beta-Globin Promoter DNA Methylation. Blood 2007, 110, 1343–1352. [Google Scholar] [CrossRef]
- Bao, X.; Zuo, Y.; Chen, D.; Zhao, C. DNA Methylation Patterns of β-Globin Cluster in β-Thalassemia Patients. Clin. Epigenetics 2020, 12, 187. [Google Scholar] [CrossRef]
- Lessard, S.; Beaudoin, M.; Benkirane, K.; Lettre, G. Comparison of DNA Methylation Profiles in Human Fetal and Adult Red Blood Cell Progenitors. Genome. Med. 2015, 7, 1. [Google Scholar] [CrossRef]
- Goren, A.; Simchen, G.; Fibach, E.; Szabo, P.E.; Tanimoto, K.; Chakalova, L.; Pfeifer, G.P.; Fraser, P.J.; Engel, J.D.; Cedar, H. Fine Tuning of Globin Gene Expression by DNA Methylation. PLoS ONE 2006, 1, e46. [Google Scholar] [CrossRef]
- Gong, Y.; Zhang, X.; Zhang, Q.; Zhang, Y.; Ye, Y.; Yu, W.; Shao, C.; Yan, T.; Huang, J.; Zhong, J.; et al. A Natural DNMT1 Mutation Elevates the Fetal Hemoglobin Level via Epigenetic Derepression of the γ-Globin Gene in β-Thalassemia. Blood 2021, 137, 1652–1657. [Google Scholar] [CrossRef]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting Histone Modifications and the Functional Organization of Mammalian Genomes. Nat. Rev. Genet. 2011, 12, 7–18. [Google Scholar] [CrossRef]
- Romano, O.; Petiti, L.; Felix, T.; Meneghini, V.; Portafax, M.; Antoniani, C.; Amendola, M.; Bicciato, S.; Peano, C.; Miccio, A. GATA Factor-Mediated Gene Regulation in Human Erythropoiesis. iScience 2020, 23, 101018. [Google Scholar] [CrossRef] [PubMed]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Rank, G.; Cerruti, L.; Simpson, R.J.; Moritz, R.L.; Jane, S.M.; Zhao, Q. Identification of a PRMT5-Dependent Repressor Complex Linked to Silencing of Human Fetal Globin Gene Expression. Blood 2010, 116, 1585–1592. [Google Scholar] [CrossRef]
- Chang, K.-H.; Fang, X.; Wang, H.; Huang, A.; Cao, H.; Yang, Y.; Bonig, H.; Stamatoyannopoulos, J.A.; Papayannopoulou, T. Epigenetic Modifications and Chromosome Conformations of the Beta Globin Locus throughout Development. Stem. Cell Rev. Rep. 2013, 9, 397–407. [Google Scholar] [CrossRef]
- Xu, J.; Shao, Z.; Glass, K.; Bauer, D.E.; Pinello, L.; Van Handel, B.; Hou, S.; Stamatoyannopoulos, J.A.; Mikkola, H.K.A.; Yuan, G.-C.; et al. Combinatorial Assembly of Developmental Stage-Specific Enhancers Controls Gene Expression Programs during Human Erythropoiesis. Dev. Cell 2012, 23, 796–811. [Google Scholar] [CrossRef]
- Hsu, M.; Richardson, C.A.; Olivier, E.; Qiu, C.; Bouhassira, E.E.; Lowrey, C.H.; Fiering, S. Complex Developmental Patterns of Histone Modifications Associated with the Human Beta-Globin Switch in Primary Cells. Exp. Hematol. 2009, 37, 799–806.e4. [Google Scholar] [CrossRef]
- Kim, A.; Kiefer, C.M.; Dean, A. Distinctive Signatures of Histone Methylation in Transcribed Coding and Noncoding Human Beta-Globin Sequences. Mol. Cell Biol. 2007, 27, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Cui, S.; Engel, J.D.; Tanabe, O. Lysine-Specific Demethylase 1 Is a Therapeutic Target for Fetal Hemoglobin Induction. Nat. Med. 2013, 19, 291–294. [Google Scholar] [CrossRef]
- Renneville, A.; Van Galen, P.; Canver, M.C.; McConkey, M.; Krill-Burger, J.M.; Dorfman, D.M.; Holson, E.B.; Bernstein, B.E.; Orkin, S.H.; Bauer, D.E.; et al. EHMT1 and EHMT2 Inhibition Induces Fetal Hemoglobin Expression. Blood 2015, 126, 1930–1939. [Google Scholar] [CrossRef]
- Krivega, I.; Byrnes, C.; de Vasconcellos, J.F.; Lee, Y.T.; Kaushal, M.; Dean, A.; Miller, J.L. Inhibition of G9a Methyltransferase Stimulates Fetal Hemoglobin Production by Facilitating LCR/γ-Globin Looping. Blood 2015, 126, 665–672. [Google Scholar] [CrossRef]
- Nualkaew, T.; Khamphikham, P.; Pongpaksupasin, P.; Kaewsakulthong, W.; Songdej, D.; Paiboonsukwong, K.; Sripichai, O.; Engel, J.D.; Hongeng, S.; Fucharoen, S.; et al. UNC0638 Induces High Levels of Fetal Hemoglobin Expression in β-Thalassemia/HbE Erythroid Progenitor Cells. Ann. Hematol. 2020, 99, 2027–2036. [Google Scholar] [CrossRef]
- Zhao, Q.; Rank, G.; Tan, Y.T.; Li, H.; Moritz, R.L.; Simpson, R.J.; Cerruti, L.; Curtis, D.J.; Patel, D.J.; Allis, C.D.; et al. PRMT5-Mediated Methylation of Histone H4R3 Recruits DNMT3A, Coupling Histone and DNA Methylation in Gene Silencing. Nat. Struct. Mol. Biol. 2009, 16, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.; Wang, Y.; Liu, R.; Zhang, Y.; Xu, Z.; Wang, Y.; Wu, Y.; Liu, M.; Cerruti, L.; Zou, F.; et al. Human Fetal Globin Gene Expression Is Regulated by LYAR. Nucleic Acids Res. 2014, 42, 9740–9752. [Google Scholar] [CrossRef] [PubMed]
- Shvedunova, M.; Akhtar, A. Modulation of Cellular Processes by Histone and Non-Histone Protein Acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- López-Bañuelos, L.; Vega, L. Inhibition of Acetylation, Is It Enough to Fight Cancer? Crit. Rev. Oncol. Hematol. 2022, 176, 103752. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, E.C.; Downs, K.M.; Christensen, H.M.; Im, H.; Nuzzi, P.A.; Bresnick, E.H. Developmentally Dynamic Histone Acetylation Pattern of a Tissue-Specific Chromatin Domain. Proc. Natl. Acad. Sci. USA 2000, 97, 14494–14499. [Google Scholar] [CrossRef]
- Im, H.; Grass, J.A.; Christensen, H.M.; Perkins, A.; Bresnick, E.H. Histone Deacetylase-Dependent Establishment and Maintenance of Broad Low-Level Histone Acetylation within a Tissue-Specific Chromatin Domain. Biochemistry 2002, 41, 15152–15160. [Google Scholar] [CrossRef]
- Schübeler, D.; Francastel, C.; Cimbora, D.M.; Reik, A.; Martin, D.I.; Groudine, M. Nuclear Localization and Histone Acetylation: A Pathway for Chromatin Opening and Transcriptional Activation of the Human Beta-Globin Locus. Genes Dev. 2000, 14, 940–950. [Google Scholar] [CrossRef]
- Yin, W.; Barkess, G.; Fang, X.; Xiang, P.; Cao, H.; Stamatoyannopoulos, G.; Li, Q. Histone Acetylation at the Human Beta-Globin Locus Changes with Developmental Age. Blood 2007, 110, 4101–4107. [Google Scholar] [CrossRef]
- Dai, Y.; Chen, T.; Ijaz, H.; Cho, E.H.; Steinberg, M.H. SIRT1 Activates the Expression of Fetal Hemoglobin Genes. Am. J. Hematol. 2017, 92, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Kim, Y.W.; Kang, J.; Kim, A. Histone H3K4me1 and H3K27ac Play Roles in Nucleosome Eviction and ERNA Transcription, Respectively, at Enhancers. FASEB J. 2021, 35, e21781. [Google Scholar] [CrossRef] [PubMed]
- Madzo, J.; Liu, H.; Rodriguez, A.; Vasanthakumar, A.; Sundaravel, S.; Caces, D.B.D.; Looney, T.J.; Zhang, L.; Lepore, J.B.; Macrae, T.; et al. Hydroxymethylation at Gene Regulatory Regions Directs Stem/Early Progenitor Cell Commitment during Erythropoiesis. Cell. Rep. 2014, 6, 231–244. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Rank, G.; Zhang, M.; Ju, J.; Liu, R.; Xu, Z.; Brown, F.; Cerruti, L.; Ma, C.; Tan, R.; et al. Induction of Human Fetal Hemoglobin Expression by Adenosine-2’,3’-Dialdehyde. J. Transl. Med. 2013, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Blobel, G.A.; Nakajima, T.; Eckner, R.; Montminy, M.; Orkin, S.H. CREB-Binding Protein Cooperates with Transcription Factor GATA-1 and Is Required for Erythroid Differentiation. Proc. Natl. Acad. Sci. USA 1998, 95, 2061–2066. [Google Scholar] [CrossRef]
- Desai, M.A.; Webb, H.D.; Sinanan, L.M.; Scarsdale, J.N.; Walavalkar, N.M.; Ginder, G.D.; Williams, D.C. An Intrinsically Disordered Region of Methyl-CpG Binding Domain Protein 2 (MBD2) Recruits the Histone Deacetylase Core of the NuRD Complex. Nucleic Acids Res. 2015, 43, 3100–3113. [Google Scholar] [CrossRef]
- Cui, S.; Kolodziej, K.E.; Obara, N.; Amaral-Psarris, A.; Demmers, J.; Shi, L.; Engel, J.D.; Grosveld, F.; Strouboulis, J.; Tanabe, O. Nuclear Receptors TR2 and TR4 Recruit Multiple Epigenetic Transcriptional Corepressors That Associate Specifically with the Embryonic β-Type Globin Promoters in Differentiated Adult Erythroid Cells▿. Mol. Cell Biol. 2011, 31, 3298–3311. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Lee, J.; Wang, H.; Miller, J.; Reik, A.; Gregory, P.D.; Dean, A.; Blobel, G.A. Controlling Long-Range Genomic Interactions at a Native Locus by Targeted Tethering of a Looping Factor. Cell 2012, 149, 1233–1244. [Google Scholar] [CrossRef]
- Guo, X.; Plank-Bazinet, J.; Krivega, I.; Dale, R.K.; Dean, A. Embryonic Erythropoiesis and Hemoglobin Switching Require Transcriptional Repressor ETO2 to Modulate Chromatin Organization. Nucleic Acids Res. 2020, 48, 10226–10240. [Google Scholar] [CrossRef]
- Saunthararajah, Y.; Hillery, C.A.; Lavelle, D.; Molokie, R.; Dorn, L.; Bressler, L.; Gavazova, S.; Chen, Y.-H.; Hoffman, R.; DeSimone, J. Effects of 5-Aza-2′-Deoxycytidine on Fetal Hemoglobin Levels, Red Cell Adhesion, and Hematopoietic Differentiation in Patients with Sickle Cell Disease. Blood 2003, 102, 3865–3870. [Google Scholar] [CrossRef]
- Park, J.W.; Han, J.-W. Targeting Epigenetics for Cancer Therapy. Arch. Pharm. Res. 2019, 42, 159–170. [Google Scholar] [CrossRef]
- Grimmer, M.R.; Stolzenburg, S.; Ford, E.; Lister, R.; Blancafort, P.; Farnham, P.J. Analysis of an Artificial Zinc Finger Epigenetic Modulator: Widespread Binding but Limited Regulation. Nucleic Acids Res. 2014, 42, 10856–10868. [Google Scholar] [CrossRef]
- Durai, S.; Mani, M.; Kandavelou, K.; Wu, J.; Porteus, M.H.; Chandrasegaran, S. Zinc Finger Nucleases: Custom-Designed Molecular Scissors for Genome Engineering of Plant and Mammalian Cells. Nucleic Acids Res. 2005, 33, 5978–5990. [Google Scholar] [CrossRef]
- Moore, M.; Klug, A.; Choo, Y. Improved DNA Binding Specificity from Polyzinc Finger Peptides by Using Strings of Two-Finger Units. Proc. Natl. Acad. Sci. USA 2001, 98, 1437–1441. [Google Scholar] [CrossRef]
- Segal, D.J.; Barbas, C.F. Custom DNA-Binding Proteins Come of Age: Polydactyl Zinc-Finger Proteins. Curr. Opin. Biotechnol. 2001, 12, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Spratt, S.K.; Liu, Q.; Johnstone, B.; Qi, H.; Raschke, E.E.; Jamieson, A.C.; Rebar, E.J.; Wolffe, A.P.; Case, C.C. Synthetic Zinc Finger Transcription Factor Action at an Endogenous Chromosomal Site. J. Biol. Chem. 2000, 275, 33850–33860. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-Q.; Rebar, E.J.; Zhang, L.; Liu, Q.; Jamieson, A.C.; Liang, Y.; Qi, H.; Li, P.-X.; Chen, B.; Mendel, M.C.; et al. Regulation of an Endogenous Locus Using a Panel of Designed Zinc Finger Proteins Targeted to Accessible Chromatin Regions. J. Biol. Chem. 2001, 276, 11323–11334. [Google Scholar] [CrossRef]
- Wilber, A.; Tschulena, U.; Hargrove, P.W.; Kim, Y.-S.; Persons, D.A.; Barbas, C.F.; Nienhuis, A.W. A Zinc-Finger Transcriptional Activator Designed to Interact with the γ-Globin Gene Promoters Enhances Fetal Hemoglobin Production in Primary Human Adult Erythroblasts. Blood 2010, 115, 3033–3041. [Google Scholar] [CrossRef]
- Huisman, C.; van der Wijst, M.G.; Falahi, F.; Overkamp, J.; Karsten, G.; Terpstra, M.M.; Kok, K.; van der Zee, A.G.; Schuuring, E.; Wisman, G.B.A.; et al. Prolonged Re-Expression of the Hypermethylated Gene EPB41L3 Using Artificial Transcription Factors and Epigenetic Drugs. Epigenetics 2015, 10, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Huisman, C.; van der Wijst, M.G.P.; Schokker, M.; Blancafort, P.; Terpstra, M.M.; Kok, K.; van der Zee, A.G.J.; Schuuring, E.; Wisman, G.B.A.; Rots, M.G. Re-Expression of Selected Epigenetically Silenced Candidate Tumor Suppressor Genes in Cervical Cancer by TET2-Directed Demethylation. Mol. Ther. 2016, 24, 536–547. [Google Scholar] [CrossRef]
- Maeder, M.L.; Angstman, J.F.; Richardson, M.E.; Linder, S.J.; Cascio, V.M.; Tsai, S.Q.; Ho, Q.H.; Sander, J.D.; Reyon, D.; Bernstein, B.E.; et al. Targeted DNA Demethylation and Activation of Endogenous Genes Using Programmable TALE-TET1 Fusion Proteins. Nat. Biotechnol. 2013, 31, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Siddique, A.N.; Nunna, S.; Rajavelu, A.; Zhang, Y.; Jurkowska, R.Z.; Reinhardt, R.; Rots, M.G.; Ragozin, S.; Jurkowski, T.P.; Jeltsch, A. Targeted Methylation and Gene Silencing of VEGF-A in Human Cells by Using a Designed Dnmt3a–Dnmt3L Single-Chain Fusion Protein with Increased DNA Methylation Activity. J. Mol. Biol. 2013, 425, 479–491. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome Editing by a CRISPR-Cas9-Based Acetyltransferase Activates Genes from Promoters and Enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Gregory, D.J.; Zhang, Y.; Kobzik, L.; Fedulov, A.V. Specific Transcriptional Enhancement of Inducible Nitric Oxide Synthase by Targeted Promoter Demethylation. Epigenetics 2013, 8, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Moreno, K.; Erices, R.; Beltran, A.S.; Stolzenburg, S.; Cuello-Fredes, M.; Owen, G.I.; Qian, H.; Blancafort, P. Breaking through an Epigenetic Wall: Re-Activation of Oct4 by KRAB-Containing Designer Zinc Finger Transcription Factors. Epigenetics 2013, 8, 164–176. [Google Scholar] [CrossRef]
- Mussolino, C.; Cathomen, T. On Target? Tracing Zinc-Finger-Nuclease Specificity. Nat. Methods 2011, 8, 725–726. [Google Scholar] [CrossRef]
- Chaikind, B.; Kilambi, K.P.; Gray, J.J.; Ostermeier, M. Targeted DNA Methylation Using an Artificially Bisected M.HhaI Fused to Zinc Fingers. PLoS ONE 2012, 7, e44852. [Google Scholar] [CrossRef]
- Joung, J.K.; Sander, J.D. TALENs: A Widely Applicable Technology for Targeted Genome Editing. Nat Rev Mol Cell Biol 2013, 14, 49–55. [Google Scholar] [CrossRef]
- Moscou, M.J.; Bogdanove, A.J. A Simple Cipher Governs DNA Recognition by TAL Effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef]
- Jankele, R.; Svoboda, P. TAL Effectors: Tools for DNA Targeting. Brief Funct. Genom. 2014, 13, 409–419. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Rogers, J.M.; Barrera, L.A.; Reyon, D.; Sander, J.D.; Kellis, M.; Keith Joung, J.; Bulyk, M.L. Context Influences on TALE–DNA Binding Revealed by Quantitative Profiling. Nat. Commun. 2015, 6, 7440. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, A.; Dubois, G.; Valton, J.; Thomas, S.; Stella, S.; Maréchal, A.; Langevin, S.; Benomari, N.; Bertonati, C.; Silva, G.H.; et al. Comprehensive Analysis of the Specificity of Transcription Activator-like Effector Nucleases. Nucleic Acids Res. 2014, 42, 5390–5402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Cong, L.; Lodato, S.; Kosuri, S.; Church, G.M.; Arlotta, P. Efficient Construction of Sequence-Specific TAL Effectors for Modulating Mammalian Transcription. Nat. Biotechnol. 2011, 29, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Yang, J.; Tsang, J.C.H.; Ooi, J.; Wu, D.; Liu, P. Reprogramming to Pluripotency Using Designer TALE Transcription Factors Targeting Enhancers. Stem Cell Rep. 2013, 1, 183–197. [Google Scholar] [CrossRef]
- Hu, J.; Lei, Y.; Wong, W.-K.; Liu, S.; Lee, K.-C.; He, X.; You, W.; Zhou, R.; Guo, J.-T.; Chen, X.; et al. Direct Activation of Human and Mouse Oct4 Genes Using Engineered TALE and Cas9 Transcription Factors. Nucleic Acids Res. 2014, 42, 4375–4390. [Google Scholar] [CrossRef]
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Dénervaud, N.; Bucher, P.; Trono, D. KRAB–Zinc Finger Proteins and KAP1 Can Mediate Long-Range Transcriptional Repression through Heterochromatin Spreading. PLoS Genet. 2010, 6, e1000869. [Google Scholar] [CrossRef]
- Cong, L.; Zhou, R.; Kuo, Y.; Cunniff, M.; Zhang, F. Comprehensive Interrogation of Natural TALE DNA-Binding Modules and Transcriptional Repressor Domains. Nat. Commun. 2012, 3, 968. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, E.; Qian, Z.; Wu, W.-S. A Multicolor Panel of TALE-KRAB Based Transcriptional Repressor Vectors Enabling Knockdown of Multiple Gene Targets. Sci. Rep. 2015, 4, 7338. [Google Scholar] [CrossRef]
- Masuda, J.; Kawamoto, H.; Strober, W.; Takayama, E.; Mizutani, A.; Murakami, H.; Ikawa, T.; Kitani, A.; Maeno, N.; Shigehiro, T.; et al. Transient Tcf3 Gene Repression by TALE-Transcription Factor Targeting. Appl. Biochem. Biotechnol. 2016, 180, 1559–1573. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Hsu, P.D.; Heidenreich, M.; Le, C.; Platt, R.J.; Scott, D.A.; Church, G.M.; Zhang, F. Optical Control of Mammalian Endogenous Transcription and Epigenetic States. Nature 2013, 500, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Ou, K.; Yu, M.; Moss, N.G.; Wang, Y.J.; Wang, A.W.; Nguyen, S.C.; Jiang, C.; Feleke, E.; Kameswaran, V.; Joyce, E.F.; et al. Targeted Demethylation at the CDKN1C/P57 Locus Induces Human β Cell Replication. J. Clin. Investig. 2018, 129, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.L.; Le Lay, J.E.; Ruano, E.G.; Kaestner, K.H. TALE-Mediated Epigenetic Suppression of CDKN2A Increases Replication in Human Fibroblasts. J. Clin. Invest. 2015, 125, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, E.M.; Williamson, K.E.; Reyon, D.; Zou, J.Y.; Ram, O.; Joung, J.K.; Bernstein, B.E. Locus-Specific Editing of Histone Modifications at Endogenous Enhancers. Nat. Biotechnol. 2013, 31, 1133–1136. [Google Scholar] [CrossRef]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232.e14. [Google Scholar] [CrossRef]
- Mlambo, T.; Nitsch, S.; Hildenbeutel, M.; Romito, M.; Müller, M.; Bossen, C.; Diederichs, S.; Cornu, T.I.; Cathomen, T.; Mussolino, C. Designer Epigenome Modifiers Enable Robust and Sustained Gene Silencing in Clinically Relevant Human Cells. Nucleic Acids Res. 2018, 46, 4456–4468. [Google Scholar] [CrossRef]
- Tesson, L.; Usal, C.; Ménoret, S.; Leung, E.; Niles, B.J.; Remy, S.; Santiago, Y.; Vincent, A.I.; Meng, X.; Zhang, L.; et al. Knockout Rats Generated by Embryo Microinjection of TALENs. Nat. Biotechnol. 2011, 29, 695–696. [Google Scholar] [CrossRef]
- Guilinger, J.P.; Pattanayak, V.; Reyon, D.; Tsai, S.Q.; Sander, J.D.; Joung, J.K.; Liu, D.R. Broad Specificity Profiling of TALENs Results in Engineered Nucleases with Improved DNA-Cleavage Specificity. Nat. Methods 2014, 11, 429–435. [Google Scholar] [CrossRef]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 Transcriptional Activators for Target Specificity Screening and Paired Nickases for Cooperative Genome Engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef]
- Polstein, L.R.; Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Bledsoe, P.; Song, L.; Safi, A.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Genome-Wide Specificity of DNA Binding, Gene Regulation, and Chromatin Remodeling by TALE- and CRISPR/Cas9-Based Transcriptional Activators. Genome Res. 2015, 25, 1158–1169. [Google Scholar] [CrossRef]
- Cuculis, L.; Zhao, C.; Abil, Z.; Zhao, H.; Shukla, D.; Schroeder, C.M. Divalent Cations Promote TALE DNA-Binding Specificity. Nucleic Acids Res. 2020, 48, 1406–1422. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA–Guided Activation of Endogenous Human Genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef]
- Moreno, A.M.; Fu, X.; Zhu, J.; Katrekar, D.; Shih, Y.-R.V.; Marlett, J.; Cabotaje, J.; Tat, J.; Naughton, J.; Lisowski, L.; et al. In Situ Gene Therapy via AAV-CRISPR-Cas9-Mediated Targeted Gene Regulation. Mol. Ther. 2018, 26, 1818–1827. [Google Scholar] [CrossRef]
- Moses, C.; Hodgetts, S.I.; Nugent, F.; Ben-Ary, G.; Park, K.K.; Blancafort, P.; Harvey, A.R. Transcriptional Repression of PTEN in Neural Cells Using CRISPR/DCas9 Epigenetic Editing. Sci. Rep. 2020, 10, 11393. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Ousterout, D.G.; Brunger, J.M.; Farin, A.M.; Glass, K.A.; Guilak, F.; Crawford, G.E.; Hartemink, A.J.; Gersbach, C.A. Synergistic and Tunable Human Gene Activation by Combinations of Synthetic Transcription Factors. Nat. Methods 2013, 10, 239–242. [Google Scholar] [CrossRef]
- Gao, X.; Tsang, J.C.H.; Gaba, F.; Wu, D.; Lu, L.; Liu, P. Comparison of TALE Designer Transcription Factors and the CRISPR/DCas9 in Regulation of Gene Expression by Targeting Enhancers. Nucleic Acids Res. 2014, 42, e155. [Google Scholar] [CrossRef]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed Activation of Endogenous Genes by CRISPR-on, an RNA-Guided Transcriptional Activator System. Cell Res. 2013, 23, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly Efficient Cas9-Mediated Transcriptional Programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lv, S.; Luo, Z.; Hu, Y.; Peng, X.; Lv, J.; Zhao, S.; Feng, J.; Huang, G.; Wan, Q.-L.; et al. MiniCAFE, a CRISPR/Cas9-Based Compact and Potent Transcriptional Activator, Elicits Gene Expression in Vivo. Nucleic Acids Res. 2021, 49, 4171–4185. [Google Scholar] [CrossRef]
- Kantor, B.; Tagliafierro, L.; Gu, J.; Zamora, M.E.; Ilich, E.; Grenier, C.; Huang, Z.Y.; Murphy, S.; Chiba-Falek, O. Downregulation of SNCA Expression by Targeted Editing of DNA Methylation: A Potential Strategy for Precision Therapy in PD. Mol. Ther. 2018, 26, 2638–2649. [Google Scholar] [CrossRef] [PubMed]
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient Targeted DNA Methylation with Chimeric DCas9–Dnmt3a–Dnmt3L Methyltransferase. Nucleic Acids Res. 2017, 45, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-Wide Programmable Transcriptional Memory by CRISPR-Based Epigenome Editing. Cell 2021, 184, 2503–2519.e17. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247.e17. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992.e6. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-DCas9 Mediated TET1 Targeting for Selective DNA Demethylation at BRCA1 Promoter. Oncotarget 2016, 7, 46545–46556. [Google Scholar] [CrossRef]
- Wilk, C.; Effenberg, L.; Abberger, H.; Steenpass, L.; Hansen, W.; Zeschnigk, M.; Kirschning, C.; Buer, J.; Kehrmann, J. CRISPR/Cas9-Mediated Demethylation of FOXP3-TSDR toward Treg-Characteristic Programming of Jurkat T Cells. Cell. Immunol. 2022, 371, 104471. [Google Scholar] [CrossRef]
- Okada, M.; Kanamori, M.; Someya, K.; Nakatsukasa, H.; Yoshimura, A. Stabilization of Foxp3 Expression by CRISPR-DCas9-Based Epigenome Editing in Mouse Primary T Cells. Epigenetics Chromatin. 2017, 10, 24. [Google Scholar] [CrossRef]
- Fang, S.; Cui, D.; Hong, T.; Guo, L.; Lee, Y.-T.; Lee, M.; Isgandarova, S.; Martinez-Moczygemba, M.; Zhou, Y.; Li, J.; et al. Ten-Eleven Translocation Ablation Impairs Cardiac Differentiation of Mouse Embryonic Stem Cells. Stem. Cells 2022, 40, 260–272. [Google Scholar] [CrossRef]
- Kabadi, A.M.; Machlin, L.; Dalal, N.; Lee, R.E.; McDowell, I.; Shah, N.N.; Drowley, L.; Randell, S.H.; Reddy, T.E. Epigenome Editing of the CFTR-Locus for Treatment of Cystic Fibrosis. J. Cyst. Fibros. 2022, 21, 164–171. [Google Scholar] [CrossRef]
- Kwon, D.Y.; Zhao, Y.-T.; Lamonica, J.M.; Zhou, Z. Locus-Specific Histone Deacetylation Using a Synthetic CRISPR-Cas9-Based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef] [PubMed]
- Kearns, N.A.; Pham, H.; Tabak, B.; Genga, R.M.; Silverstein, N.J.; Garber, M.; Maehr, R. Functional Annotation of Native Enhancers with a Cas9–Histone Demethylase Fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef]
- Williams, R.M.; Senanayake, U.; Artibani, M.; Taylor, G.; Wells, D.; Ahmed, A.A.; Sauka-Spengler, T. Genome and Epigenome Engineering CRISPR Toolkit for in Vivo Modulation of Cis -Regulatory Interactions and Gene Expression in the Chicken Embryo. Development 2018, 145, dev.160333. [Google Scholar] [CrossRef] [PubMed]
- O’Geen, H.; Ren, C.; Nicolet, C.M.; Perez, A.A.; Halmai, J.; Le, V.M.; Mackay, J.P.; Farnham, P.J.; Segal, D.J. DCas9-Based Epigenome Editing Suggests Acquisition of Histone Methylation Is Not Sufficient for Target Gene Repression. Nucleic Acids Res. 2017, 45, 9901–9916. [Google Scholar] [CrossRef] [PubMed]
- O’Geen, H.; Tomkova, M.; Combs, J.A.; Tilley, E.K.; Segal, D.J. Determinants of Heritable Gene Silencing for KRAB-DCas9 + DNMT3 and Ezh2-DCas9 + DNMT3 Hit-and-Run Epigenome Editing. Nucleic Acids Res. 2022, 50, 3239–3253. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-Throughput Profiling of off-Target DNA Cleavage Reveals RNA-Programmed Cas9 Nuclease Specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-Fidelity CRISPR–Cas9 Nucleases with No Detectable Genome-Wide off-Target Effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-J.; Orlova, N.; Oakes, B.L.; Ma, E.; Spinner, H.B.; Baney, K.L.M.; Chuck, J.; Tan, D.; Knott, G.J.; Harrington, L.B.; et al. CasX Enzymes Comprise a Distinct Family of RNA-Guided Genome Editors. Nature 2019, 566, 218–223. [Google Scholar] [CrossRef]
- Kiani, S.; Chavez, A.; Tuttle, M.; Hall, R.N.; Chari, R.; Ter-Ovanesyan, D.; Qian, J.; Pruitt, B.W.; Beal, J.; Vora, S.; et al. Cas9 GRNA Engineering for Genome Editing, Activation and Repression. Nat. Methods 2015, 12, 1051–1054. [Google Scholar] [CrossRef]
- Liu, J.; Sun, M.; Cho, K.B.; Gao, X.; Guo, B. A CRISPR-Cas9 Repressor for Epigenetic Silencing of KRAS. Pharmacol. Res. 2021, 164, 105304. [Google Scholar] [CrossRef]
- Zheng, Y.; Shen, W.; Zhang, J.; Yang, B.; Liu, Y.-N.; Qi, H.; Yu, X.; Lu, S.-Y.; Chen, Y.; Xu, Y.-Z.; et al. CRISPR Interference-Based Specific and Efficient Gene Inactivation in the Brain. Nat. Neurosci. 2018, 21, 447–454. [Google Scholar] [CrossRef]
- Mussolino, C. Precise Epigenome Editing on the Stage: A Novel Approach to Modulate Gene Expression. Epigenet. Insights 2018, 11, 2516865718818838. [Google Scholar] [CrossRef]
- Kiefer, C.M.; Hou, C.; Little, J.A.; Dean, A. Epigenetics of Beta-Globin Gene Regulation. Mutat. Res. 2008, 647, 68–76. [Google Scholar] [CrossRef]
- Kalantri, S.A.; Ray, R.; Chattopadhyay, A.; Bhattacharjee, S.; Biswas, A.; Bhattacharyya, M. Efficacy of Decitabine as Hemoglobin F Inducer in HbE/β-Thalassemia. Ann. Hematol. 2018, 97, 1689–1694. [Google Scholar] [CrossRef]
- Molokie, R.; Lavelle, D.; Gowhari, M.; Pacini, M.; Krauz, L.; Hassan, J.; Ibanez, V.; Ruiz, M.A.; Ng, K.P.; Woost, P.; et al. Oral Tetrahydrouridine and Decitabine for Non-Cytotoxic Epigenetic Gene Regulation in Sickle Cell Disease: A Randomized Phase 1 Study. PLoS Med. 2017, 14, e1002382. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, A.G.; Groy, A.; Gore, E.R.; Atkins, C.; Long, E.R.; Montoute, M.N.; Wu, Z.; Halsey, W.; McNulty, D.E.; Ennulat, D.; et al. In Vitro and in Vivo Induction of Fetal Hemoglobin with a Reversible and Selective DNMT1 Inhibitor. Haematologica 2021, 106, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Okam, M.M.; Esrick, E.B.; Mandell, E.; Campigotto, F.; Neuberg, D.S.; Ebert, B.L. Phase 1/2 Trial of Vorinostat in Patients with Sickle Cell Disease Who Have Not Benefitted from Hydroxyurea. Blood 2015, 125, 3668–3669. [Google Scholar] [CrossRef] [PubMed]
- Junker, L.H.; Li, B.; Zhu, X.; Koti, S.; Cerbone, R.E.; Hendrick, C.L.; Sangerman, J.; Perrine, S.; Pace, B.S. Novel Histone Deacetylase Inhibitor CT-101 Induces γ-Globin Gene Expression in Sickle Erythroid Progenitors with Targeted Epigenetic Effects. Blood Cells Mol. Dis. 2022, 93, 102626. [Google Scholar] [CrossRef]
- Cui, S.; Lim, K.-C.; Shi, L.; Lee, M.; Jearawiriyapaisarn, N.; Myers, G.; Campbell, A.; Harro, D.; Iwase, S.; Trievel, R.C.; et al. The LSD1 Inhibitor RN-1 Induces Fetal Hemoglobin Synthesis and Reduces Disease Pathology in Sickle Cell Mice. Blood 2015, 126, 386–396. [Google Scholar] [CrossRef]
- Rivers, A.; Vaitkus, K.; Ruiz, M.A.; Ibanez, V.; Jagadeeswaran, R.; Kouznetsova, T.; DeSimone, J.; Lavelle, D. RN-1, a Potent and Selective Lysine-Specific Demethylase 1 Inhibitor, Increases γ-Globin Expression, F Reticulocytes, and F Cells in a Sickle Cell Disease Mouse Model. Exp. Hematol. 2015, 43, 546–553.e1–3. [Google Scholar] [CrossRef] [PubMed]
- Rivers, A.; Vaitkus, K.; Ibanez, V.; Ruiz, M.A.; Jagadeeswaran, R.; Saunthararajah, Y.; Cui, S.; Engel, J.D.; DeSimone, J.; Lavelle, D. The LSD1 Inhibitor RN-1 Recapitulates the Fetal Pattern of Hemoglobin Synthesis in Baboons (P. Anubis). Haematologica 2016, 101, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Matson, D.; Xie, K.; Roth, M.; Stuart, B.; Bruno, P.; Efremov, I.; Thompson, L.; Silver, S.; Moxham, C. Ftx-6058 Induces Fetal Hemoglobin Production and Ameliorates Disease Pathology in Sickle Cell Mice. Blood 2021, 138, 2018. [Google Scholar] [CrossRef]
- Sher, F.; Hossain, M.; Seruggia, D.; Schoonenberg, V.A.C.; Yao, Q.; Cifani, P.; Dassama, L.M.K.; Cole, M.A.; Ren, C.; Vinjamur, D.S.; et al. Rational Targeting of a NuRD Subcomplex Guided by Comprehensive in Situ Mutagenesis. Nat. Genet. 2019, 51, 1149–1159. [Google Scholar] [CrossRef]
- Ginder, G.D. Epigenetic Regulation of Fetal Globin Gene Expression in Adult Erythroid Cells. Transl. Res. 2015, 165, 115–125. [Google Scholar] [CrossRef]
- Amendola, M.; Brusson, M.; Miccio, A. CRISPRthripsis: The Risk of CRISPR/Cas9-Induced Chromothripsis in Gene Therapy. Stem Cells Transl. Med. 2022, 11, 1003–1009. [Google Scholar] [CrossRef]
- Costa, F.C.; Fedosyuk, H.; Neades, R.; de Los Rios, J.B.; Barbas, C.F.; Peterson, K.R. Induction of Fetal Hemoglobin In Vivo Mediated by a Synthetic γ -Globin Zinc Finger Activator. Anemia 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly Specific Epigenome Editing by CRISPR-Cas9 Repressors for Silencing of Distal Regulatory Elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liu, Y.; Cao, H.; Zhang, Y.; Gu, Z.; Liu, X.; Yu, A.; Kaphle, P.; Dickerson, K.E.; Ni, M.; et al. Interrogation of Enhancer Function by Enhancer-Targeting CRISPR Epigenetic Editing. Nat. Commun. 2020, 11, 485. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontana, L.; Alahouzou, Z.; Miccio, A.; Antoniou, P. Epigenetic Regulation of β-Globin Genes and the Potential to Treat Hemoglobinopathies through Epigenome Editing. Genes 2023, 14, 577. https://doi.org/10.3390/genes14030577
Fontana L, Alahouzou Z, Miccio A, Antoniou P. Epigenetic Regulation of β-Globin Genes and the Potential to Treat Hemoglobinopathies through Epigenome Editing. Genes. 2023; 14(3):577. https://doi.org/10.3390/genes14030577
Chicago/Turabian StyleFontana, Letizia, Zoe Alahouzou, Annarita Miccio, and Panagiotis Antoniou. 2023. "Epigenetic Regulation of β-Globin Genes and the Potential to Treat Hemoglobinopathies through Epigenome Editing" Genes 14, no. 3: 577. https://doi.org/10.3390/genes14030577
APA StyleFontana, L., Alahouzou, Z., Miccio, A., & Antoniou, P. (2023). Epigenetic Regulation of β-Globin Genes and the Potential to Treat Hemoglobinopathies through Epigenome Editing. Genes, 14(3), 577. https://doi.org/10.3390/genes14030577