

The Telomeric Repeats of HHV-6A Do Not Determine the Chromosome into Which the Virus Is Integrated

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Search
2.2. Alignment
2.3. Identification and Visualisation of Indels
2.4. Search for Tandem Repeats
3. Results
3.1. Search Results for HHV-6A Sequences
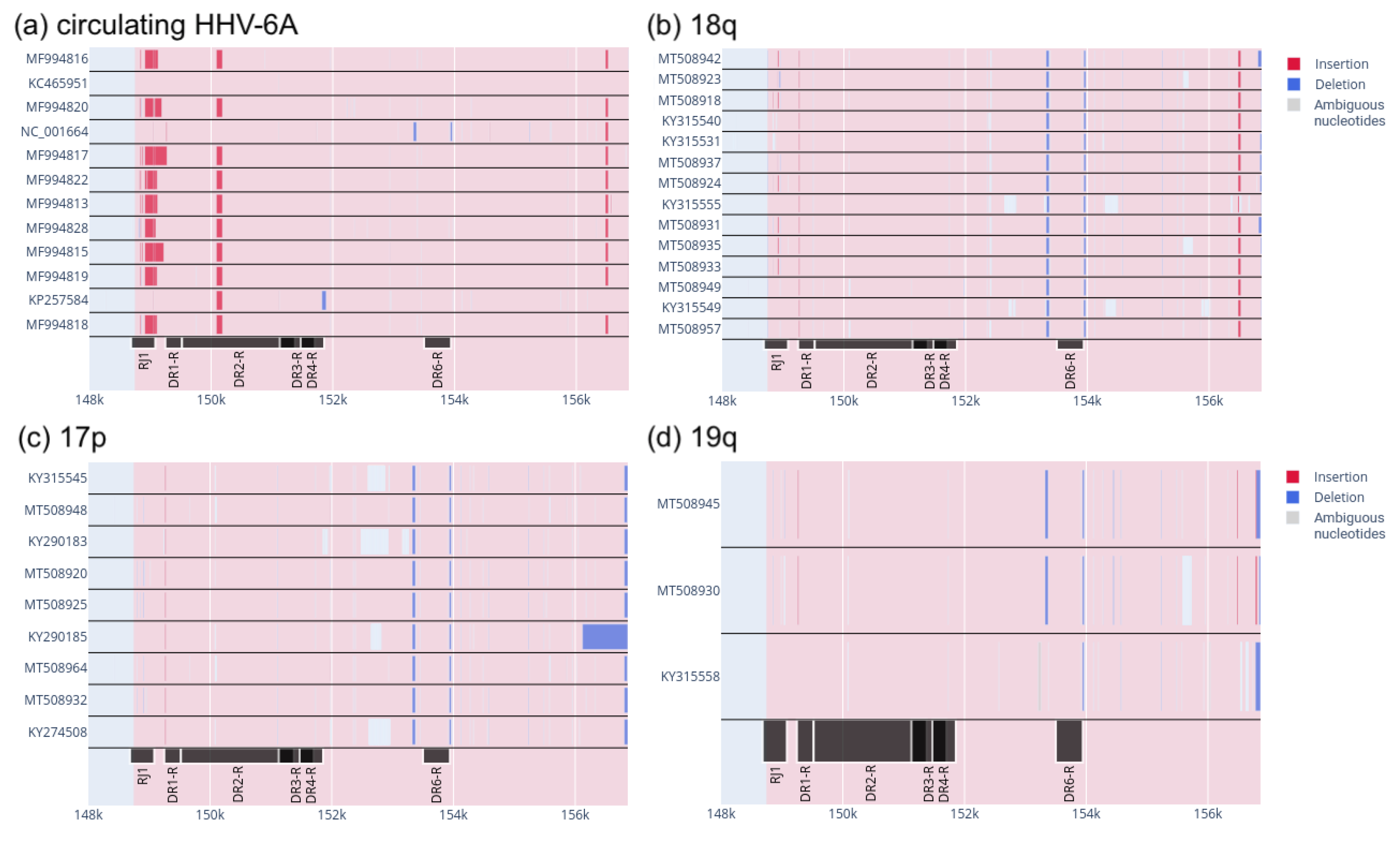
3.2. Patterns of Insertions and Deletions in the DRR
3.3. TMR Does Not Identify the Chromosome for HHV-6A Integration
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- King, O.; Khalili, Y.A. Herpes Virus Type 6; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Agut, H.; Bonnafous, P.; Gautheret-Dejean, A. Update on infections with human herpes viruses 6A, 6B, and 7. Médecine Mal. Infect. 2017, 47, 83–91. [Google Scholar] [CrossRef]
- Dominguez, G.; Dambaugh, T.R.; Stamey, F.R.; Dewhurst, S.; Inoue, N.; Pellett, P.E. Human Herpes virus 6B Genome Sequence: Coding Content and Comparison with Human Herpes virus 6A. J. Virol. 1999, 73, 8040–8052. [Google Scholar] [CrossRef]
- Gompels, U.A.; Kasolo, F.C. HHV-6 Genome: Similar and Different; Elsevier: Amsterdam, The Netherlands, 2006; Volume 12, pp. 23–46. [Google Scholar] [CrossRef]
- Achour, A.; Malet, I.; Deback, C.; Bonnafous, P.; Boutolleau, D.; Gautheret-Dejean, A.; Agut, H. Length variability of telomeric repeat sequences of human herpes virus 6 DNA. J. Virol. Methods 2009, 159, 127–130. [Google Scholar] [CrossRef]
- Luppi, M.; Marasca, R.; Barozzi, P.; Ferrari, S.; Ceccherini-Nelli, L.; Batoni, G.; Merelli, E.; Torelli, G. Three cases of human herpes virus-6 latent infection: Integration of viral genome in peripheral blood mononuclear cell DNA. J. Med. Virol. 1993, 40, 44–52. [Google Scholar] [CrossRef]
- Daibata, M.; Taguchi, T.; Nemoto, Y.; Taguchi, H.; Miyoshi, I. Inheritance of Chromosomally Integrated Human Herpes virus 6 DNA. Blood 1999, 94, 1545–1549. [Google Scholar] [CrossRef]
- Nacheva, E.P.; Ward, K.N.; Brazma, D.; Virgili, A.; Howard, J.; Leong, H.N.; Clark, D.A. Human herpes virus 6 integrates within telomeric regions as evidenced by five different chromosomal sites. J. Med. Virol. 2008, 80, 1952–1958. [Google Scholar] [CrossRef]
- Clark, D.A.; Nacheva, E.P.; Leong, H.N.; Brazma, D.; Li, Y.T.; Tsao, E.H.F.; Buyck, H.C.E.; Atkinson, C.E.; Lawson, H.M.; Potter, M.N.; et al. Transmission of Integrated Human Herpes virus 6 through Stem Cell Transplantation: Implications for Laboratory Diagnosis. J. Infect. Dis. 2006, 193, 912–916. [Google Scholar] [CrossRef]
- Hubacek, P.; Virgili, A.; Ward, K.N.; Pohlreich, D.; Keslova, P.; Goldova, B.; Markova, M.; Zajac, M.; Cinek, O.; Nacheva, E.P. HHV-6 DNA throughout the tissues of two stem cell transplant patients with chromosomally integrated HHV-6 and fatal CMV pneumonitis. Br. J. Haematol. 2009, 145, 394–398. [Google Scholar] [CrossRef]
- Ohye, T.; Kawamura, Y.; Inagaki, H.; Yoshikawa, A.; Ihira, M.; Yoshikawa, T.; Kurahashi, H. A simple cytogenetic method to detect chromosomally integrated human herpes virus-6. J. Virol. Methods 2016, 228, 74–78. [Google Scholar] [CrossRef]
- Miura, H.; Kawamura, Y.; Hattori, F.; Kozawa, K.; Ihira, M.; Ohye, T.; Kurahashi, H.; Yoshikawa, T. Chromosomally integrated human herpes virus 6 in the Japanese population. J. Med. Virol. 2018, 90, 1636–1642. [Google Scholar] [CrossRef]
- Greninger, A.L.; Naccache, S.N.; Pannaraj, P.; Jerome, K.R.; Dien, B.J.; Ruderman, J.W.; Burnham, C.A.D. The Brief Case: Inherited Chromosomally Integrated Human Herpes virus 6 (HHV-6) in the Age of Multiplex HHV-6 Testing. J. Clin. Microbiol. 2022, 57, e02016-18. [Google Scholar] [CrossRef] [PubMed]
- Kheimar, A.; Previdelli, R.L.; Wight, D.J.; Kaufer, B.B. Telomeres and telomerase: Role in Marek’s disease virus pathogenesis, integration and tumorigenesis. Viruses 2017, 9, 173. [Google Scholar] [CrossRef] [PubMed]
- Bertzbach, L.D.; Kheimar, A.; Ali, F.A.Z.; Kaufer, B.B. Viral Factors Involved in Marek’s Disease Virus (MDV) Pathogenesis. Curr. Clin. Microbiol. Rep. 2018, 5, 238–244. [Google Scholar] [CrossRef]
- Eliassen, E.; Krueger, G.; Luppi, M.; Ablashi, D. Lymphoproliferative Syndromes Associated with Human Herpes virus-6A and Human Herpes virus-6B. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018035. [Google Scholar] [CrossRef]
- Kühl, U.; Lassner, D.; Wallaschek, N.; Gross, U.M.; Krueger, G.R.F.; Seeberg, B.; Kaufer, B.B.; Escher, F.; Poller, W.; Schultheiss, H.P. Chromosomally integrated human herpes virus 6 in heart failure: Prevalence and treatment. Eur. J. Heart Fail. 2015, 17, 9–19. [Google Scholar] [CrossRef]
- Aimola, G.; Beythien, G.; Aswad, A.; Kaufer, B.B. Current understanding of human herpes virus 6 (HHV-6) chromosomal integration. Antivir. Res. 2020, 176, 104720. [Google Scholar] [CrossRef]
- Wallaschek, N.; Sanyal, A.; Pirzer, F.; Gravel, A.; Mori, Y.; Flamand, L.; Kaufer, B.B. The Telomeric Repeats of Human Herpes virus 6A (HHV-6A) Are Required for Efficient Virus Integration. PLoS Pathog. 2016, 12, e1005666. [Google Scholar] [CrossRef]
- Aswad, A.; Aimola, G.; Wight, D.; Roychoudhury, P.; Zimmermann, C.; Hill, J.; Lassner, D.; Xie, H.; Huang, M.L.; Parrish, N.F.; et al. Evolutionary History of Endogenous Human Herpes virus 6 Reflects Human Migration out of Africa. Mol. Biol. Evol. 2021, 38, 96–107. [Google Scholar] [CrossRef]
- Gompels, U.A.; Nicholas, J.; Lawrence, G.; Jones, M.; Thomson, B.J.; Martin, M.E.D.; Efstathiou, S.; Craxton, M.; Macaulay, H.A. The DNA Sequence of Human Herpes virus-6: Structure, Coding Content, and Genome Evolution. Virology 1995, 209, 29–51. [Google Scholar] [CrossRef]
- Gravel, A.; Sinnett, D.; Flamand, L. Frequency of Chromosomally-Integrated Human Herpes virus 6 in Children with Acute Lymphoblastic Leukemia. PLoS ONE 2013, 8, e84322. [Google Scholar] [CrossRef]
- Tweedy, J.; Spyrou, M.A.; Donaldson, C.D.; Depledge, D.; Breuer, J.; Gompels, U.A. Complete Genome Sequence of the Human Herpes virus 6A Strain AJ from Africa Resembles Strain GS from North America. Genome Announc. 2015, 3, e01498-14. [Google Scholar] [CrossRef]
- Tweedy, J.; Spyrou, M.A.; Pearson, M.; Lassner, D.; Kuhl, U.; Gompels, U.A. Complete genome sequence of germline chromosomally integrated human herpes virus 6A and analyses integration sites define a new human endogenous virus with potential to reactivate as an emerging infection. Viruses 2016, 8, 19. [Google Scholar] [CrossRef]
- Zhang, E.; Cotton, V.E.; Hidalgo-Bravo, A.; Huang, Y.; Bell, A.J.; Jarrett, R.F.; Wilkie, G.S.; Davison, A.J.; Nacheva, E.P.; Siebert, R.; et al. HHV-8-unrelated primary effusion-like lymphoma associated with clonal loss of inherited chromosomally-integrated human herpes virus-6A from the telomere of chromosome 19q. Sci. Rep. 2016, 6, 22730. [Google Scholar] [CrossRef]
- Zhang, E.; Bell, J.A.; Wilkie, S.G.; Suárez, M.N.; Batini, C.; Colin, D.V.; Armendáriz-Castillo, I.; Neumann, R.; Victoria, E.C.; Huang, Y.; et al. Inherited Chromosomally Integrated Human Herpes virus 6 Genomes Are Ancient, Intact, and Potentially Able To Reactivate from Telomeres. J. Virol. 2017, 91, e01137-17. [Google Scholar] [CrossRef]
- Telford, M.; Navarro, A.; Santpere, G. Whole genome diversity of inherited chromosomally integrated HHV-6 derived from healthy individuals of diverse geographic origin. Sci. Rep. 2018, 8, 3472. [Google Scholar] [CrossRef]
- Greninger, A.L.; Knudsen, G.M.; Roychoudhury, P.; Hanson, D.J.; Sedlak, R.H.; Xie, H.; Guan, J.; Nguyen, T.; Peddu, V.; Boeckh, M.; et al. Comparative genomic, transcriptomic, and proteomic reannotation of human herpes virus 6. BMC Genom. 2018, 19, 204. [Google Scholar] [CrossRef]
- Greninger, L.A.; Roychoudhury, P.; Makhsous, N.; Hanson, D.; Chase, J.; Krueger, G.; Xie, H.; Huang, M.L.; Saunders, L.; Ablashi, D.; et al. Copy Number Heterogeneity, Large Origin Tandem Repeats, and Interspecies Recombination in Human Herpes virus 6A (HHV-6A) and HHV-6B Reference Strains. J. Virol. 2018, 92, e00135-18. [Google Scholar] [CrossRef]
- Wood, M.L.; Veal, C.D.; Neumann, R.; Suárez, N.M.; Nichols, J.; Parker, A.J.; Martin, D.; Romaine, S.P.R.; Codd, V.; Samani, N.J.; et al. Variation in human herpes virus 6B telomeric integration, excision, and transmission between tissues and individuals. eLife 2021, 10, e70452. [Google Scholar] [CrossRef]
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.; Dempsey, D.; Dickerman, A.; Dietrich, E.; Kenyon, R.; et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): A resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2022, 51, D678–D689. [Google Scholar] [CrossRef]
- Peddu, V.; Greninger, A.; Roychoudhury, P. Direct Submission. In NCBI Nucleotide Accession Number MH698400 (Submitted 27-JUL-2018), MH698403 (Submitted 30-JUL-2018); Laboratory Medicine, Virology, University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- Domonova, E.A.; Goptar, I.A.; Nikiforova, A.V.; Kuleshov, K.V. Direct Submission. In NCBI Nucleotide Accession Number MK630133 and MK630134 (Submitted 14-MAR-2019); Laboratory of Enteric Diseases Epidemiology, Central Research Institute for Epidemiology: Moscow, Russia, 2019. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Istvan, A. Available online: https://github.com/ialbert/bio (accessed on 25 November 2022).
- Plotly Technologies Inc. Collaborative Data Science. Available online: https://plot.ly (accessed on 28 November 2022).
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Loewenthal, G.; Rapoport, D.; Avram, O.; Moshe, A.; Wygoda, E.; Itzkovitch, A.; Israeli, O.; Azouri, D.; Cartwright, R.A.; Mayrose, I.; et al. A Probabilistic Model for Indel Evolution: Differentiating Insertions from Deletions. Mol. Biol. Evol. 2021, 38, 5769–5781. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clade | NCBI ID |
|---|---|
| Circulating viruses | KJ123690, NC_001664, KP257584, MF994822, KC465951, MF994820, MF994815, MF994816, MF994817, MF994818, MF994819, MF994828, MF994813. |
| “17p” clade | KJ123690, KY316055, KY316048, KY290185, KY274508, KY290183, KY315545, MW049318, MW049313, MW049315, MW049316, MW049322, MK630133, MK630134, MT508920, MT508925, MT508932, MT508948, MT508964. |
| “18q” clade | KJ123690, KY315531, KY315540, KY315549, KY315555, MW049314, MT508918, MT508923, MT508924, MT508931, MT508933, MT508935, MT508937, MT508942, MT508949, MT508957. |
| “19q” clade | KJ123690, KT355575, KY316054, MG894371, KY315558, MW049317, MW049319, MW049320, MW049321, MT508930, MT508945. |
| Clade | NCBI ID |
|---|---|
| Circulating viruses | KJ123690, NC_001664, KP257584, MF994822, KC465951, MF994820, MF994815, MF994816, MF994817, MF994818, MF994819, MF994828, MF994813. |
| “17p” clade | MT508948, MT508964. |
| “18q” clade | MT508918, MT508923, MT508949, MT508957. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kusakin, A.V.; Goleva, O.V.; Danilov, L.G.; Krylov, A.V.; Tsay, V.V.; Kalinin, R.S.; Tian, N.S.; Eismont, Y.A.; Mukomolova, A.L.; Chukhlovin, A.B.; et al. The Telomeric Repeats of HHV-6A Do Not Determine the Chromosome into Which the Virus Is Integrated. Genes 2023, 14, 521. https://doi.org/10.3390/genes14020521
Kusakin AV, Goleva OV, Danilov LG, Krylov AV, Tsay VV, Kalinin RS, Tian NS, Eismont YA, Mukomolova AL, Chukhlovin AB, et al. The Telomeric Repeats of HHV-6A Do Not Determine the Chromosome into Which the Virus Is Integrated. Genes. 2023; 14(2):521. https://doi.org/10.3390/genes14020521
Chicago/Turabian StyleKusakin, Aleksey V., Olga V. Goleva, Lavrentii G. Danilov, Andrey V. Krylov, Victoria V. Tsay, Roman S. Kalinin, Natalia S. Tian, Yuri A. Eismont, Anna L. Mukomolova, Alexei B. Chukhlovin, and et al. 2023. "The Telomeric Repeats of HHV-6A Do Not Determine the Chromosome into Which the Virus Is Integrated" Genes 14, no. 2: 521. https://doi.org/10.3390/genes14020521
APA StyleKusakin, A. V., Goleva, O. V., Danilov, L. G., Krylov, A. V., Tsay, V. V., Kalinin, R. S., Tian, N. S., Eismont, Y. A., Mukomolova, A. L., Chukhlovin, A. B., Komissarov, A. S., & Glotov, O. S. (2023). The Telomeric Repeats of HHV-6A Do Not Determine the Chromosome into Which the Virus Is Integrated. Genes, 14(2), 521. https://doi.org/10.3390/genes14020521