

Homozygous Missense Variant in the Solute Carrier Organic Anion Transporter 2A1 (SLCO2A1) Gene Underlies Isolated Nail Clubbing



Abstract
1. Introduction
2. Materials and Methods
2.1. Approval and DNA Extraction
2.2. Whole Exome Sequencing (WES)
2.3. Validation and Segregation Analysis by Sanger Sequencing
2.4. In Silico Tools
2.5. 3D Protein Modeling
3. Results
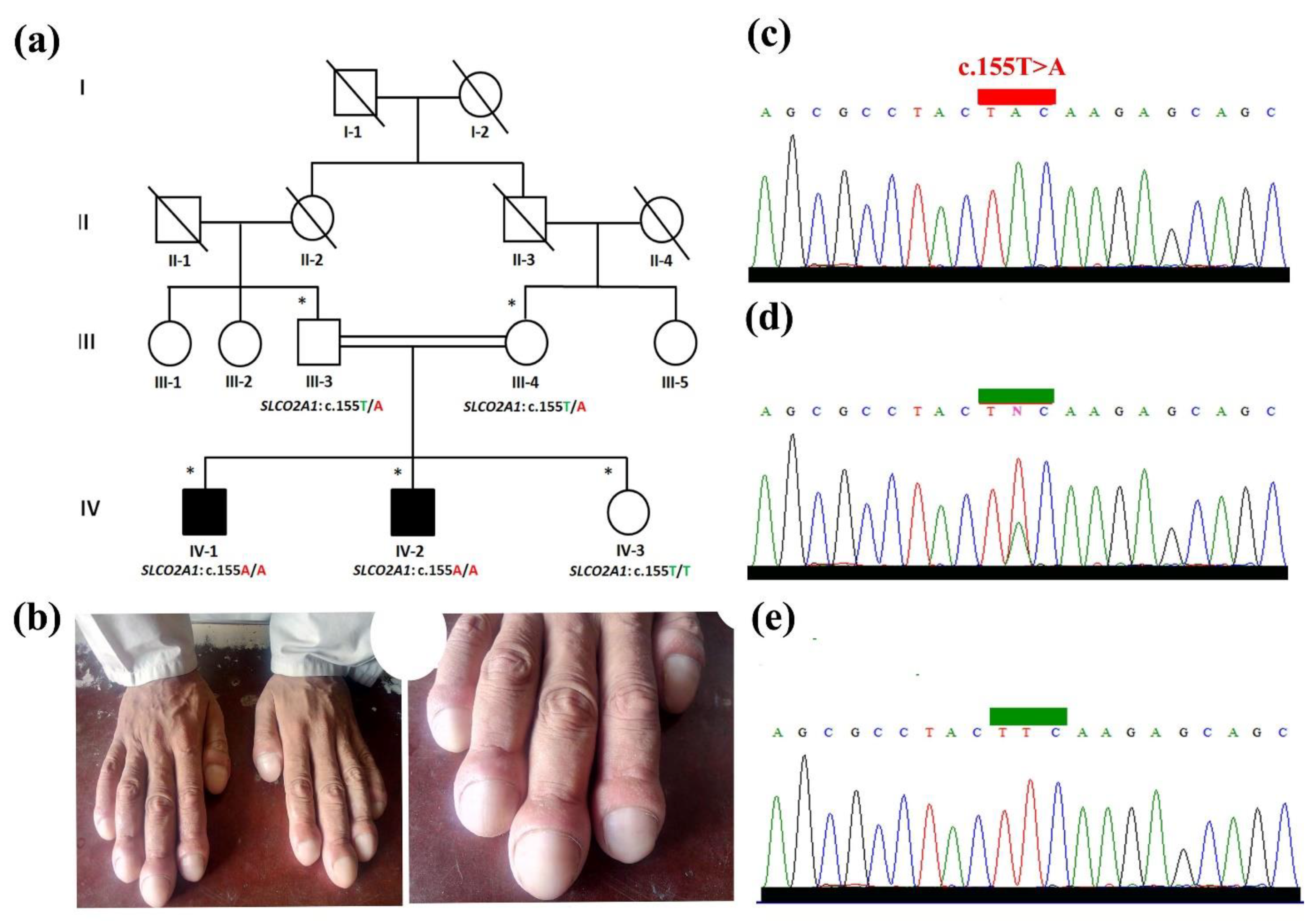
3.1. Clinical Evaluation
3.2. Whole Exome Data Analysis
3.3. Validation and Segregation Analysis
3.4. 3D Protein Modeling of SLCO2A1 (Phe52Tyr)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sarkar, M.; Mahesh, D.M.; Madabhavi, I. Digital clubbing. Lung India 2012, 29, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xia, W.; He, J.; Zhang, Z.; Ke, Y.; Yue, H.; Wang, C.; Zhang, H.; Gu, J.; Hu, W.; et al. Exome sequencing identifies SLCO2A1 mutations as a cause of primary hypertrophic osteoarthropathy. Am. J. Hum. Genet. 2012, 90, 125–132. [Google Scholar] [CrossRef]
- Burcovschii, S.; Aboeed, A. Nail Clubbing; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Vandemergel, X.; Renneboog, B. Prevalence, etiology and significance of clubbing in a Department of General Internal Medicine. Eur. J. Intern. Med. 2008, 19, 325–329. [Google Scholar]
- Goodyer, M.J.; Cronin, M.; Ketsitlile, D.G.; O’Reilly, S.P.; Moylan, E.J. Hodgkin’s lymphoma with digital clubbing. J. Clin. Oncol. 2009, 27, e95–e96. [Google Scholar] [CrossRef]
- Nadrous, H.F.; Myers, J.L.; Decker, P.A.; Ryu, J.H. Idiopathic pulmonary fibrosis in patients younger than 50 years. Mayo Clin. Proc. 2005, 80, 37–40. [Google Scholar] [PubMed]
- Seifert, W.; Kühnisch, J.; Tüysüz, B.; Specker, C.; Brouwers, A.; Horn, D. Mutations in the prostaglandin transporter encoding gene SLCO2A1 cause primary hypertrophic osteoarthropathy and isolated digital clubbing. Hum. Mutat. 2012, 33, 660–664. [Google Scholar] [PubMed]
- Tariq, M.; Azeem, Z.; Ali, G.; Chishti, M.S.; Ahmad, W. Mutation in the HPGD gene encoding NAD+ dependent 15-hydroxyprostaglandin dehydrogenase underlies isolated congenital nail clubbing (ICNC). J. Med. Genet. 2009, 46, 14–20. [Google Scholar] [PubMed]
- Erken, E.; Köroğlu, Ç.; Yıldız, F.; Özer, H.T.; Gülek, B.; Tolun, A. A novel recessive 15-hydroxyprostaglandin dehydrogenase mutation in a family with primary hypertrophic osteoarthropathy. Mod. Rheumatol. 2015, 25, 315–321. [Google Scholar]
- Ullah, A.; Gul, A.; Umair, M.; Irfanullah.; Ahmad, F.; Aziz, A.; Wali, A.; Ahmad, W. Homozygous sequence variants in the WNT10B gene underlie split hand/foot malformation. Genet. Mol. Biol. 2018, 41, 1–8. [Google Scholar] [CrossRef]
- Umair, M.; Ahmad, F.; Ahmad, S.; Alam, Q.; Rehan, M.; Alqosaibi, A.I.; Alnamshan, M.M.; Rafeeq, M.M.; Haque, S.; Sain, Z.M.; et al. A Novel Homozygous Missense Mutation in the Zinc Finger DNA Binding Domain of GLI1 Causes Recessive Post-Axial Polydactyly. Front. Genet. 2021, 12, 746949. [Google Scholar]
- Rafeeq, M.M.; Umair, M.; Bilal, M.; Habib, A.H.; Waqas, A.; Sain, Z.M.; Alam, M.Z.; Ali, R.H. A novel biallelic variant further delineates PRDX3-related autosomal recessive cerebellar ataxia. Neurogenetics 2022, 24, 55–60. [Google Scholar] [CrossRef]
- Waqas, A.; Nayab, A.; Shaheen, S.; Abbas, S.; Latif, M.; Rafeeq, M.M.; Al-Dhuayan, I.S.; Alqosaibi, A.I.; Alnamshan, M.M.; Sain, Z.M.; et al. Case Report: Biallelic Variant in the tRNA Methyltransferase Domain of the AlkB Homolog 8 Causes Syndromic Intellectual Disability. Front. Genet. 2022, 13, 878274. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.; Samad, A.; Shah, A.A.; Wadood, A.; Alkathiri, A.; Alshehri, M.A.; Alam, M.Z.; Hussain, T.; He, P.; Umair, M. A novel biallelic variant in the Popeye domain-containing protein 1 (POPDC1) underlies limb girdle muscle dystrophy type 25. Clin. Genet. 2022, 103, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Nayab, A.; Alam, Q.; Alzahrani, O.R.; Khan, R.; Sarfaraz, S.; Albaz, A.A.; Rafeeq, M.M.; Sain, Z.M.; Waqas, A.; Umair, M. Targeted exome sequencing identified a novel frameshift variant in the PGAM2 gene causing glycogen storage disease type X. Eur. J. Med. Genet. 2021, 64, 104283. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Umair, M.; Majeed, A.I.; Abdullah; Jan, A.; Ahmad, W. A novel homozygous sequence variant in GLI1 underlies first case of autosomal recessive pre-axial polydactyly. Clin. Genet. 2019, 95, 540–541. [Google Scholar]
- Myers, K.A.; Farquhar, D.R. The rational clinical examination. Does this patient have clubbing? JAMA 2001, 286, 341–347. [Google Scholar] [CrossRef]
- Dubrey, S.; Pal, S.; Singh, S.; Karagiannis, G. Digital clubbing: Forms, associations and pathophysiology. Br. J. Hosp. Med. 2016, 77, 403–408. [Google Scholar]
- Shah, K.; Ferrara, T.; Jan, A.; Umair, M.; Irfanullah; Khan, S.; Ahmad, W.; Spritz, R. Homozygous SLCO 2A1 translation initiation codon mutation in a Pakistani family with recessive isolated congenital nail clubbing. Br. J. Dermatol. 2017, 177, 546–548. [Google Scholar] [CrossRef]
- Diggle, C.P.; Parry, D.A.; Logan, C.V.; Laissue, P.; Rivera, C.; Restrepo, C.M.; Fonseca, D.J.; Morgan, J.E.; Allanore, Y.; Fontenay, M.; et al. Prostaglandin transporter mutations cause pachydermoperiostosis with myelofibrosis. Hum. Mutat. 2012, 33, 1175–1181. [Google Scholar] [CrossRef]
- Zhang, Z.; He, J.W.; Fu, W.Z.; Zhang, C.Q.; Zhang, Z.L. A novel mutation in the SLCO2A1 gene in a Chinese family with primary hypertrophic osteoarthropathy. Gene 2013, 521, 191–194. [Google Scholar]
- Sasaki, T.; Niizeki, H.; Shimizu, A.; Shiohama, A.; Hirakiyama, A.; Okuyama, T.; Seki, A.; Kabashima, K.; Otsuka, A.; Ishiko, A.; et al. Identification of mutations in the prostaglandin transporter gene SLCO2A1 and its phenotype-genotype correlation in Japanese patients with pachydermoperiostosis. J. Dermatol. Sci. 2012, 68, 36–44. [Google Scholar] [PubMed]
- Zhang, Z.; He, J.-W.; Fu, W.-Z.; Zhang, C.-Q.; Zhang, Z.-L. Mutations in the SLCO2A1 gene and primary hypertrophic osteoarthropathy: A clinical and biochemical characterization. J. Clin. Endocrinol. Metab. 2013, 98, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Kanai, N.; Bao, Y.; Schuster, V.L. Cloning, in vitro expression, and tissue distribution of a human prostaglandin transporter cDNA(hPGT). J. Clin. Invest. 1996, 98, 1142–1149. [Google Scholar] [PubMed]
- Schuster, V.L. Prostaglandin transport. Prostaglandins Other Lipid Mediat. 2002, 68–69, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Lu, R.; Pucci, M.L.; Schuster, V.L. The two-step model of prostaglandin signal termination: In vitro reconstitution with the prostaglandin transporter and prostaglandin 15 dehydrogenase. Mol. Pharmacol. 2004, 65, 973–978. [Google Scholar]
- Endo, S.; Nomura, T.; Chan, B.S.; Lu, R.; Pucci, M.L.; Bao, Y.; Schuster, V.L. Expression of PGT in MDCK cell monolayers: Polarized apical localization and induction of active PG transport. Am. J. Physiol. Renal. Physiol. 2002, 282, 618–622. [Google Scholar]
- Coggins, K.G.; Coffman, T.M.; Koller, B.H. The Hippocratic finger points the blame at PGE2. Nat. Genet. 2008, 40, 691–692. [Google Scholar]
- Bloch, A.; Couture, G.; Isidor, B.; Ricquebourg, M.; Bourrat, E.; Lipsker, D.; Taillan, B.; Combier, A.; Chiaverini, C.; Moufle, F.; et al. Novel pathogenic variants in SLCO2A1 causing autosomal dominant primary hypertrophic osteoarthropathy. EJMG 2023, 66, 104689. [Google Scholar] [CrossRef]
- Li, S.S.; He, J.W.; Fu, W.Z.; Liu, Y.J.; Hu, Y.Q.; Zhang, Z.L. Clinical, Biochemical, and Genetic Features of 41 Han Chinese Families with Primary Hypertrophic Osteoarthropathy, and Their Therapeutic Response to Etoricoxib: Results from a Six-Month Prospective Clinical Intervention. J. Bone Min. Res. 2017, 32, 1659–1666. [Google Scholar] [CrossRef]
- Chang, H.Y.; Locker, J.; Lu, R.; Schuster, V.L. Failure of postnatal ductus arteriosus closure in prostaglandin transporter-deficient mice. Circulation 2010, 121, 529–536. [Google Scholar] [CrossRef]
- Guda, K.; Fink, S.P.; Milne, G.L.; Molyneaux, N.; Ravi, L.; Lewis, S.M.; Dannenberg, A.J.; Montgomery, C.G.; Zhang, S.; Willis, J.; et al. Inactivating mutation in the prostaglandin transporter gene, SLCO2A1, associated with familial digital clubbing, colon neoplasia, and NSAID resistance. Cancer Prev. Res. 2014, 7, 805–812. [Google Scholar]
- Niizeki, H.; Shiohama, A.; Sasaki, T.; Seki, A.; Kabashima, K.; Otsuka, A.; Kosaki, K.; Ogo, A.; Yamada, T.; Miyasaka, M.; et al. The complete type of pachydermoperiostosis: A novel nonsense mutation p.E141* of the SLCO2A1 gene. J. Dermatol. Sci. 2014, 75, 193–195. [Google Scholar] [PubMed]
- Uppal, S.; Diggle, C.P.; Carr, I.M.; Fishwick, C.W.G.; Ahmed, M.; Ibrahim, G.H.; Helliwell, P.S.; Latos-Bieleńska, A.; Phillips, S.E.V.; Markham, A.F.; et al. Mutations in 15-hydroxyprostaglandin dehydrogenase cause primary hypertrophic osteoarthropathy. Nat. Genet. 2008, 40, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Van Poucke, M.; Melkebeek, V.; Erkens, T.; Van Zeveren, A.; Cox, E.; Peelman, L.J. Molecular cloning and characterization of the porcine prostaglandin transporter (SLCO2A1): Evaluation of its role in F4mediated neonatal diarrhoea. BMC Genet. 2009, 10, 64. [Google Scholar]
- Alfadhel, M.; Umair, M.; Almuzzaini, B.; Alsaif, S.; AlMohaimeed, S.A.; Almashary, M.A.; Alharbi, W.; Alayyar, L.; Alasiri, A.; Ballow, M.; et al. Targeted SLC19A3 gene sequencing of 3000 Saudi newborn: A pilot study toward newborn screening. Ann. Clin. Transl. Neurol. 2019, 6, 2097–2103. [Google Scholar] [CrossRef] [PubMed]
- Alyafee, Y.; Alam, Q.; Tuwaijri, A.; Umair, M.; Haddad, S.; Alharbi, M.; Alrabiah, H.; Al-Ghuraibi, M.; Al-Showaier, S.; Alfadhel, M. Next-Generation Sequencing-Based Pre-Implantation Genetic Testing for Aneuploidy (PGT-A): First Report from Saudi Arabia. Genes 2021, 12, 461. [Google Scholar] [CrossRef]
- Alyafee, Y.; Al Tuwaijri, A.; Alam, Q.; Umair, M.; Haddad, S.; Alharbi, M.; Ballow, M.; Al Drees, M.; AlAbdulrahman, A.; Al Khaldi, A.; et al. Next Generation Sequencing Based Non-invasive Prenatal Testing (NIPT): First Report from Saudi Arabia. Front. Genet. 2021, 12, 630787. [Google Scholar] [CrossRef]
- Alyafee, Y.; Al Tuwaijri, A.; Umair, M.; Alharbi, M.; Haddad, S.; Ballow, M.; Alayyar, L.; Alam, Q.; Althenayyan, S.; Al Ghilan, N.; et al. Non-invasive Prenatal Testing for Autosomal Recessive Disorders: A New Promising Approach. Front. Genet. 2022, 13, 1047474. [Google Scholar] [CrossRef]
- Ahmad, F.; Ali, R.H.; Muhammad, D.; Nasir, A.; Umair, M.; Wakil, S.M.; Ramzan, K.; Basit, S.; Ahmad, W. Novel homozygous sequence variants in the CDH3 gene encoding P-cadherin underlying hypotrichosis with juvenile macular dystrophy in consanguineous families. Eur. J. Dermatol. 2016, 26, 610–612. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Phenotypic/Genotypic Characteristics | Family Members | ||||
|---|---|---|---|---|---|
| VI-1 | VI-2 | VI-3 | III-3 | III-4 | |
| Sex | Male | Male | Female | Male | Female |
| Consanguinity | + | + | + | + | + |
| Clinical subtype | Complete | Complete | N.A | N.A | N.A |
| Mutation type | Homozygous Missense | Homozygous Missense | Homozygous Normal | Heterozygous carrier | Heterozygous carrier |
| SLCO2A1 allele 1 | c.155T>A | c.155T>A | c.155T>T | c.155T>A | c.155T>A |
| SLCO2A1 allele 2 | c.155T>A | c.155T>A | c.155T>T | c.155T>T | c.155T>T |
| Protein change | p.Phe52Tyr | p.Phe52Tyr | N.A | N.A | N.A |
| Digital clubbing | + | + | − | − | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umair, M.; Bilal, M.; Shah, K.; Said, G.; Ahmad, F. Homozygous Missense Variant in the Solute Carrier Organic Anion Transporter 2A1 (SLCO2A1) Gene Underlies Isolated Nail Clubbing. Genes 2023, 14, 430. https://doi.org/10.3390/genes14020430
Umair M, Bilal M, Shah K, Said G, Ahmad F. Homozygous Missense Variant in the Solute Carrier Organic Anion Transporter 2A1 (SLCO2A1) Gene Underlies Isolated Nail Clubbing. Genes. 2023; 14(2):430. https://doi.org/10.3390/genes14020430
Chicago/Turabian StyleUmair, Muhammad, Muhammad Bilal, Khadim Shah, Gulab Said, and Farooq Ahmad. 2023. "Homozygous Missense Variant in the Solute Carrier Organic Anion Transporter 2A1 (SLCO2A1) Gene Underlies Isolated Nail Clubbing" Genes 14, no. 2: 430. https://doi.org/10.3390/genes14020430
APA StyleUmair, M., Bilal, M., Shah, K., Said, G., & Ahmad, F. (2023). Homozygous Missense Variant in the Solute Carrier Organic Anion Transporter 2A1 (SLCO2A1) Gene Underlies Isolated Nail Clubbing. Genes, 14(2), 430. https://doi.org/10.3390/genes14020430