
Excluding Digenic Inheritance of PGAP2 and PGAP3 Variants in Mabry Syndrome (OMIM 239300) Patient: Phenotypic Spectrum Associated with PGAP2 Gene Variants in Hyperphosphatasia with Mental Retardation Syndrome-3 (HPMRS3)


 ,
,
Abstract
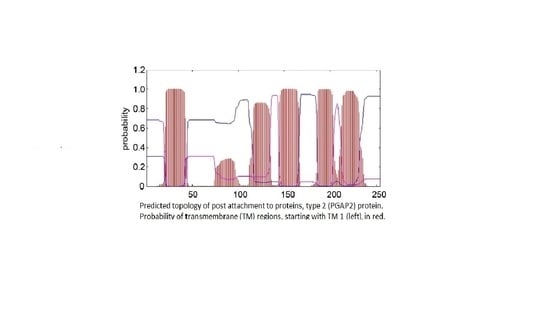
1. Introduction
2. Case Report
3. Materials and Methods
3.1. Molecular Genetics
3.2. Targeted Exome Panel Sequencing
3.3. In Vitro Rescue Assays
4. Results
4.1. Molecular Genetics
4.2. Sequencing
4.3. In Vitro Rescue Assays
5. Discussion
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mabry, C.C.; Bautista, A.; Kirk, R.F.; Dubilier, L.D.; Braunstein, H.; Koepke, J.A. Familial hyperphosphatase with mental retardation, seizures, and neurologic deficits. J. Pediatr. 1970, 77, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.D.; Nezarati, M.M.; Gillessen-Kaesbach, G.; Meinecke, P.; Mendoza, R.; Mornet, E.; Brun-Heath, I.; Squarcioni, C.P.; Legea-Mallet, L.; Munnich, A.; et al. Hyperphosphatasia with seizures, neurologic deficit, and characteristic facial features: Five new patients with Mabry syndrome. Am. J. Med. Genet. 2010, 152A, 1661–1669. [Google Scholar]
- Cole, D.E.; Thompson, M.D. Neurogenetic aspects of hyperphosphatasia in Mabry syndrome. Subcell. Biochem. 2015, 76, 343–361. [Google Scholar] [PubMed]
- Carmody, L.C.; Blau, H.; Danis, D.; Zhang, X.A.; Gourdine, J.-P.; Vasilevsky, N.; Krawitz, P.; Thompson, M.D.; Robinson, P.N. Significantly different clinical phenotypes associated with mutations in synthesis and transamidase + remodeling glycosylphosphatidylinositol (GPI)-anchor biosynthesis genes. Orphanet J. Rare Dis. 2020, 15, 40. [Google Scholar] [CrossRef]
- Krawitz, P.; Schweiger, M.R.; Rödelsperger, C.; Marcelis, C.; Kölsch, U.; Meisel, C.; Stephani, F.; Kinoshita, T.; Murakami, Y.; Bauer, S.; et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat. Genet. 2010, 42, 827–829. [Google Scholar] [CrossRef]
- Knaus, A.; Pantel, J.T.; Pendziwiat, M.; Hajjir, N.; Zhao, M.; Hsieh, T.-C.; Schubach, M.; Gurovich, Y.; Fleischer, N.; Jäger, M.; et al. Characterization of glycosylphosphatidylinositol biosynthesis defects by clinical features, flow cytometry, and automated image analysis. Genome Med. 2018, 10, 3. [Google Scholar] [CrossRef]
- Ng, G.G.; Freeze, H.H. Human genetic disorders involving glycosylphosphatidylinositol (GPI) anchors and glycosphingolipids (GSL). J. Inherit. Metab. Dis. 2015, 38, 171–178. [Google Scholar] [CrossRef]
- Thompson, M.D.; Cole, D.E. Recessive PIGN Mutations in Fryns Syndrome: Evidence for Genetic Heterogeneity. Hum. Mutat. 2016, 37, 621. [Google Scholar] [CrossRef]
- Bellai-Dussault, K.; Nguyen, T.T.M.; Baratang, V.N.; Jimenez-Cruz, D.A.; Campeau, P.M. Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders. Clin. Genet. 2019, 95, 112–121. [Google Scholar] [CrossRef]
- Kinoshita, T.; Fujita, M. Biosynthesis of GPI-anchored proteins: Special emphasis on GPI lipid remodeling. J. Lipid Res. 2016, 57, 6–24. [Google Scholar] [CrossRef]
- Murakami, Y.; Nguyen, T.T.M.; Baratang, N.; Raju, P.K.; Knaus, A.; Ellard, S.; Jones, G.; Lace, B.; Rousseau, J.; Ajeawung, N.F.; et al. Mutations in PIGB cause an inherited GPI biosynthesis defect with an axonal neuropathy and metabolic abnormality in severe cases. Am. J. Hum. Genet. 2019, 105, 384–394. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Murakami, Y.; Wigby, K.M.; Baratang, N.V.; Rousseau, J.; St-Denis, A.; Rosenfeld, J.A.; Laniewski, S.C.; Jones, J.; Iglesias, A.D.; et al. Mutations in PIGS, encoding a GPI transamidase, cause a neurological syndrome ranging from fetal akinesia to epileptic encephalopathy. Am. J. Hum. Genet. 2018, 103, 602–611. [Google Scholar] [CrossRef]
- Horn, D.; Schottmann, G.; Meinecke, P. Hyperphosphatasia with mental retardation, brachytelephalangy, and a distinct facial gestalt: Delineation of a recognizable syndrome. Eur. J. Med. Genet. 2010, 53, 85–88. [Google Scholar] [CrossRef]
- Thompson, M.D.; Roscioli, T.; Marcelis, C.; Nezarati, M.M.; Stolte-Dijkstra, I.; Sharom, F.J.; Lu, P.; Phillips, J.A.; Sweeney, E.; Robinson, P.N.; et al. Phenotypic variability in hyperphosphatasia with seizures and neurologic deficit (Mabry syndrome). Am. J. Med. Genet. 2012, 158A, 553–558. [Google Scholar] [CrossRef]
- Thompson, M.D.; Killoran, A.; Percy, M.E.; Nezarati, M.M.; Cole, D.E.; Hwang, P.A. Hyperphosphatasia with neurologic deficit: A pyridoxine-responsive seizure disorder? Pediatr. Neurol. 2006, 34, 303–307. [Google Scholar] [CrossRef]
- Xue, J.; Li, H.; Zhang, Y.; Yang, Z. Clinical and genetic analysis of two Chinese infants with Mabry syndrome. Brain Dev. 2016, 38, 807–818. [Google Scholar] [CrossRef]
- Krawitz, P.M.; Murakami, Y.; Hecht, J.; Krüger, U.; Holder, S.E.; Mortier, G.R.; Delle Chiaie, B.; De Baere, E.; Thompson, M.D.; Roscioli, T.; et al. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am. J. Hum. Genet. 2012, 91, 146–151. [Google Scholar] [CrossRef]
- Nakamura, K.; Osaka, H.; Murakami, Y.; Anzai, R.; Nishiyama, K.; Kodera, H.; Nakashima, M.; Tsurusaki, Y.; Miyake, N.; Kinoshita, T.; et al. PIGO mutations in intractable epilepsy and severe developmental delay with mild elevation of alkaline phosphatase levels. Epilepsia 2014, 55, e13–e17. [Google Scholar] [CrossRef]
- Chiyonobu, T.; Inoue, N.; Morimoto, M.; Kinoshita, T.; Murakami, Y. Glycosylphosphatidylinositol (GPI) anchor deficiency caused by mutations in PIGW is associated with West syndrome and hyperphosphatasia with mental retardation syndrome. J. Med. Genet. 2014, 51, 203–207. [Google Scholar] [CrossRef]
- Ilkovski, B.; Pagnamenta, A.J.; O’Grady, G.L.; Kinoshita, T.; Howard, M.F.; Lek, M.; Thomas, B.; Turner, A.; Christodoulou, J.; Sillence, D.; et al. Mutations in PIGY: Expanding the phenotype of inherited glycosylphosphatidylinositol deficiencies. Hum. Mol. Genet. 2015, 24, 6146–6159. [Google Scholar] [CrossRef]
- Hansen, L.; Tawamie, H.; Murakami, Y.; Mang, Y.; ur Rehman, S.; Buchert, R.; Schaffer, S.; Muhammad, S.; Bak, M.; Nöthen, M.M.; et al. Hypomorphic mutations in PGAP2, encoding a GPI-anchor-remodeling protein, cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2013, 92, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Naseer, M.I.; Rasool, M.; Jan, M.M.; Chaudhary, A.G.; Pushparaj, P.N.; Abuzenadah, A.M.; Al-Qahtani, M.H. A novel mutation in PGAP2 gene causes developmental delay, intellectual disability, epilepsy and microcephaly in consanguineous Saudi family. J. Neurol. Sci. 2016, 371, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Krawitz, P.M.; Murakami, Y.; Rie, A.; Hietala, M.; Krüger, U.; Zhu, N.; Kinoshita, T.; Mundlos, S.; Hecht, J.; Robinson, P.N.; et al. PGAP2 mutations, affecting the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation syndrome. Am. J. Hum. Genet. 2013, 92, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Jezela-Stanek, A.; Ciara, E.; Piekutowska-Abramczuk, D.; Trubicka, J.; Jurkiewicz, E.; Rokicki, D.; Mierzewska, H.; Spychalska, J.; Uhrynowska, M.; Szwarc-Bronikowska, M.; et al. Congenital disorder of glycosylphosphatidylinositol (GPI)-anchor biosynthesis—The phenotype of two patients with novel mutations in the PIGN and PGAP2 genes. Eur. J. Paediatr. Neurol. 2016, 20, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.F.; Murakami, Y.; Pagnamenta, A.T.; Daumer-Haas, C.; Fischer, B.; Hecht, J.; Keays, D.A.; Knight, S.J.; Kölsch, U.; Krüger, U.; et al. Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation. Am. J. Hum. Genet. 2014, 94, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Knaus, A.; Awaya, T.; Helbig, I.; Afawi, Z.; Pendziwiat, M.; Abu-Rachma, J.; Thompson, M.D.; Cole, D.E.; Skinner, S.; Annese, F.; et al. Rare noncoding mutations extend the mutational spectrum in the PGAP3 subtype of hyperphosphatasia with mental retardation syndrome. Hum. Mutat. 2016, 37, 737–744. [Google Scholar] [CrossRef]
- Thompson, M.D.; Cole, D.E.; Mabry, C.C. 50 Years Ago in The Journal of Pediatrics: Familial Hyperphosphatasia with Mental Retardation, Seizures, and Neurologic Deficits. J. Pediatr. 2020, 222, 97. [Google Scholar] [CrossRef]
- Thompson, M.D.; Knaus, A.A.; Barshop, B.A.; Caliebe, A.; Muhle, H.; Nguyen, T.T.M.; Baratang, N.V.; Kinoshita, T.; Percy, M.E.; Campeau, P.M.; et al. A post glycosylphosphatidylinositol (GPI) attachment to proteins, type 2 (PGAP2) variant identified in Mabry syndrome index cases: Molecular genetics of the prototypical inherited GPI disorder. Eur. J. Med. Genet. 2020, 63, 103822. [Google Scholar] [CrossRef]
- Tashima, Y.; Taguchi, R.; Murata, C.; Ashida, H.; Kinoshita, T.; Maeda, Y. PGAP2 is essential for correct processing and stable expression of GPI-anchored proteins. Mol. Biol. Cell 2006, 17, 1410–1420. [Google Scholar] [CrossRef]
- Maeda, Y.; Tashima, Y.; Houjou, T.; Fujita, M.; Yoko-o, T.; Jigami, Y.; Taguchi, R.; Kinoshita, T. Fatty acid remodeling of GPI-anchored proteins is required for their raft association. Mol. Biol. Cell 2007, 18, 1497–1506. [Google Scholar] [CrossRef]
- Chander, V.; Mahmoud, M.; Hu, J.; Dardas, Z.; Grochowski, C.M.; Dawood, M.; Khayat, M.M.; Li, H.; Li, S.; Jhangiani, S.; et al. Long read sequencing and expression studies of AHDC1 deletions in Xia-Gibbs syndrome reveal a novel genetic regulatory mechanism. Hum. Mutat. 2022, 4, 2033–2053. [Google Scholar] [CrossRef]
- Huynh, M.T.; Tran, C.T.; Joubert, M.; Bénéteau, C. Intragenic Deletion of the ZMYND11 Gene in 10p15.3 is Associated with Developmental Delay Phenotype: A Case Report. Cytogenet. Genome Res. 2021, 161, 445–448. [Google Scholar] [CrossRef]
- De Luca, O.; Salerno, G.; De Bernardini, D.; Torre, M.S.; Simmaco, M.; Lionetto, L.; Gentile, G.; Borro, M. Predicting Dihydropyrimidine Dehydrogenase Deficiency and Related 5-Fluorouracil Toxicity: Opportunities and Challenges of DPYD Exon Sequencing and the Role of Phenotyping Assays. Int. J. Mol. Sci. 2022, 23, 13923. [Google Scholar] [CrossRef]
- Friedman, J.; Feigenbaum, A.; Chuang, N.; Silhavy, J.; Gleeson, J.G. Pyruvate dehydrogenase complex-E2 deficiency causes paroxysmal exercise-induced dyskinesia. Neurology 2017, 89, 2297–2298. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | +/− |
|---|---|
| Hyperphosphatasia | + |
| Global developmental/intellectual disability | + |
| Delayed speech | + |
| Delayed age at walking/no walking | + |
| Hypotonia | + |
| Seizures | + |
| Brachytelephalangy | − |
| Tented upper lip vermillion | − |
| Broad nasal bridge | − |
| Broad nasal tip | + |
| Multiple congenital anomalies | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, M.D.; Li, X.; Spencer-Manzon, M.; Andrade, D.M.; Murakami, Y.; Kinoshita, T.; Carpenter, T.O. Excluding Digenic Inheritance of PGAP2 and PGAP3 Variants in Mabry Syndrome (OMIM 239300) Patient: Phenotypic Spectrum Associated with PGAP2 Gene Variants in Hyperphosphatasia with Mental Retardation Syndrome-3 (HPMRS3). Genes 2023, 14, 359. https://doi.org/10.3390/genes14020359
Thompson MD, Li X, Spencer-Manzon M, Andrade DM, Murakami Y, Kinoshita T, Carpenter TO. Excluding Digenic Inheritance of PGAP2 and PGAP3 Variants in Mabry Syndrome (OMIM 239300) Patient: Phenotypic Spectrum Associated with PGAP2 Gene Variants in Hyperphosphatasia with Mental Retardation Syndrome-3 (HPMRS3). Genes. 2023; 14(2):359. https://doi.org/10.3390/genes14020359
Chicago/Turabian StyleThompson, Miles D., Xueying Li, Michele Spencer-Manzon, Danielle M. Andrade, Yoshiko Murakami, Taroh Kinoshita, and Thomas O. Carpenter. 2023. "Excluding Digenic Inheritance of PGAP2 and PGAP3 Variants in Mabry Syndrome (OMIM 239300) Patient: Phenotypic Spectrum Associated with PGAP2 Gene Variants in Hyperphosphatasia with Mental Retardation Syndrome-3 (HPMRS3)" Genes 14, no. 2: 359. https://doi.org/10.3390/genes14020359
APA StyleThompson, M. D., Li, X., Spencer-Manzon, M., Andrade, D. M., Murakami, Y., Kinoshita, T., & Carpenter, T. O. (2023). Excluding Digenic Inheritance of PGAP2 and PGAP3 Variants in Mabry Syndrome (OMIM 239300) Patient: Phenotypic Spectrum Associated with PGAP2 Gene Variants in Hyperphosphatasia with Mental Retardation Syndrome-3 (HPMRS3). Genes, 14(2), 359. https://doi.org/10.3390/genes14020359