Abstract
Chromosomal rearrangements involving the KMT2A gene occur frequently in acute lymphoblastic leukaemia (ALL). KMT2A-rearranged ALL (KMT2Ar ALL) has poor long-term survival rates and is the most common ALL subtype in infants less than 1 year of age. KMT2Ar ALL frequently occurs with additional chromosomal abnormalities including disruption of the IKZF1 gene, usually by exon deletion. Typically, KMT2Ar ALL in infants is accompanied by a limited number of cooperative le-sions. Here we report a case of aggressive infant KMT2Ar ALL harbouring additional rare IKZF1 gene fusions. Comprehensive genomic and transcriptomic analyses were performed on sequential samples. This report highlights the genomic complexity of this particular disease and describes the novel gene fusions IKZF1::TUT1 and KDM2A::IKZF1.
1. Introduction
The histone-lysine [K] Methyl Transferase 2A (KMT2A) on chromosome 11q23 is a pathogenic driver gene in acute lymphoblastic leukemia (ALL). KMT2A-rearranged ALL (KMT2Ar ALL) has poor long-term survival rates and is the most common ALL subtype in infants (<1 year of age) [1], comprising >70% of all ALL diagnoses in this age group [2]. It is often associated with hyperleukocytosis and central nervous system (CNS) involvement [3]. While KMT2Ar ALL frequently occurs with additional chromosomal abnormalities including disruption of the IKZF1 gene (chromosome 7p12), usually by exon deletion [4], KMT2Ar ALL in infants typically presents with few co-occurring alterations [5]. However, we report a genomically complex case of aggressive congenital KMT2Ar ALL harboring additional rare and novel IKZF1 gene fusions.
2. Case Report
A full-term neonate was delivered to a mother with an unremarkable antenatal history. At three days old the infant had a complete blood count performed in the setting of hyperbilirubinemia and poor feeding. This revealed a leukocytosis of 136 × 109/L (98% lymphoblasts), with normal red cell and platelet parameters (hemoglobin 171 × 109/L, platelets 183 × 109/L). Immunophenotyping of peripheral blood mononuclear cells (PBMNC) confirmed B-ALL (70% blasts; 93% CD19/CD34+, 39% CD10/CD19/CD34+). Cytogenetics demonstrated a 47XX karyotype with trisomy 8 and translocations t(4;11)(q21;q23) and t(7;11)(q11.2;p11.2). The KMT2A::AFF1 (MLL::MLLT2/AF4) KMT2A rearrangement was confirmed by fluorescence in situ hybridization. No diagnostic bone marrow biopsy was performed. A lumbar puncture revealed CNS involvement.
The infant was treated with induction chemotherapy as per the Interfant-06 protocol [6] and achieved a minimal residual disease (MRD) level of 5 × 10−3 by the end of induction using Allele Specific Oligonucleotide PCR for IgH rearrangement. However, further intensive chemotherapy was delayed by significant toxicities, including vincristine-related polyneuropathy requiring mechanical ventilation. Following two weeks of dose-reduced mercaptopurine and oral methotrexate maintenance, azacitidine was administered (2.5 mg/kg for 5 days) [7] with the intention of bridging to hematopoietic stem cell transplant if remission was obtained. Unfortunately, the bone marrow blast percentage rose to 5% at the end of this cycle, and despite further azacitidine and consolidation chemotherapy as per the protocol AALL15P1 [7], by the next bone marrow examination, lymphoblasts had risen to 55%. Subsequent blinatumomab combined with intrathecal chemotherapy resulted in a morphological remission with MRD of 10−2. However, the second blinatumomab course was complicated by the development of facial nerve palsy secondary to an extradural leukemic chloroma impinging on her facial nerve. This heralded florid morphological relapse with 42% bone marrow blasts, which exhibited complete loss of CD19 on flow cytometry (93% CD19/34+ at diagnosis). The patient failed to respond to either FLAG-IDA or inotuzumab and succumbed at nine months of age. The timeline of treatments and response assessments are summarized in Figure 1, and immunophenotyping flow cytometric dot plots are shown in Figure S1.
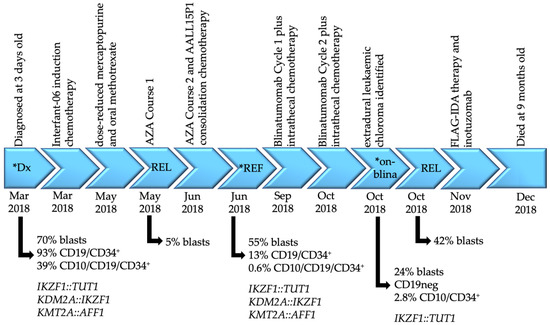
Figure 1.
Disease timeline of patient CHI_0391. Disease timepoints, associated treatments, blast percentages with the corresponding immunophenotype where available and gene fusions identified by mRNA-Seq are indicated. Asterisks denote the samples analyzed by transcriptomic sequencing in this report. Interfant-06 induction chemotherapy includes prednisone, dexamethasone, vincristine, cytarabine, daunorubicin, PEG-asparaginase, methotrexate, bortezomib and melphalan; AALL15P1 consolidation protocol includes cyclophosphamide, mercaptopurine, cytarabine, methotrexate, hydrocortisone and methotrexate. Dx = diagnosis; REF = refractory; REL = relapse; AZA = azacytidine; blina = blinatumomab; FLAG-IDA = fludarabine, cytarabine, granulocyte-colony stimulating factor, idarubicin.
3. Results
Transcriptomic sequencing (mRNA-Seq) performed on PBMNCs at diagnosis identified the KMT2A::AFF1 gene fusion, with low number of reads. Two IKZF1 gene fusions were also identified: IKZF1::TUT1 and KDM2A::IKZF1 (Figure 2A, Table S2). Both IKZF1 fusions were validated by PCR and Sanger sequencing (Figure 2B). Significantly, this is the first time the KDM2A::IKZF1 and IKZF1::TUT1 gene fusions have been described, and the first report of KMT2Ar ALL with co-occurring IKZF1 fusions [8]. Although KDM2A and TUT1 are both in the same karyotypic region of chromosome 11 (11q13.2 and 11q12.3, respectively), they are separated by >190 genes, with a genetic distance corresponding to a recombination frequency of >3.8% [9]. Thus, these two fusions likely represent separate genomic events. Multiplex ligation-dependent probe amplification (MLPA) is a PCR-based method for quantification of DNA copy numbers and a reliable method for copy number variation (CNV) genotyping. We used two different MLPA probe mixes (P202 and P335, MRC Holland) to determine CNV in genomic DNA. No deletions or duplications were detected in any of the genes assayed; of importance, no deletions were detected in IKZF1 exons 1–8 (Figure S2).
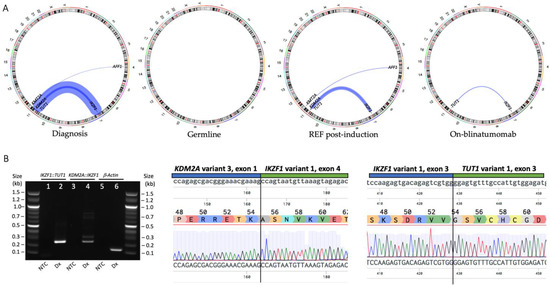
Figure 2.
(A) Circos plot representation of gene fusions identified by transcriptomic sequencing of PBMNC at diagnosis, MSC representing a germline sample, BMMNC from a refractory sample after commencing induction therapy and BMMNC from a sample while undergoing blinatumomab therapy. (B) The IKZF1::TUT1 and KDM2A::IKZF1 gene fusions were detected by breakpoint RT-PCR in the diagnosis PBMNC sample. IKZF1::TUT1 product size = 261 bp, KDM2A::IKZF1 product size = 256 bp, β-Actin = 193 bp. The fusions were validated by Sanger sequencing. The fusion breakpoints are shown in the upper panels and delineated with a vertical line, the amino acid reference sequences in the middle panels and the sequencing trace in the lower panels. Sequencing was aligned with Benchling software. Abbreviations: PBMNC = peripheral blood mononuclear cells; MSC = mesenchymal stem cells; BMMNC = bone marrow mononuclear cells; REF = refractory; Dx = diagnosis; NTC = no template control.
The KMT2A::AFF1 gene fusion observed here is the most common fusion observed in infant disease (49% of all infant KMT2Ar leukemias [1]). However, the breakpoint in KMT2A exon 9 (Figure 3A) is rarely observed in infant KMT2A::AFF1 leukemia (19%, compared with the frequently observed exon 11 breakpoint) [8]. Upon formation of KMT2A::AFF1, the entire C-terminal portion of KMT2A is lost; this region contains domains important for post-translational regulation and mediation of protein–protein interactions. The lost regulatory and H3K4 methyltransferase activity lead to the widespread epigenetic dysregulation observed in KMT2Ar patients [1]. The KMT2A portion retained in the fusion harbors binding motifs for proteins, such as menin and LEDGF, which are critical for leukemic transformation [10,11], and domains to facilitate KMT2A’s DNA-binding capacity. The AFF1 portion of the fusion lacks a degron sequence, likely perturbing the protein’s degradation rate. This is supported by high AFF1 expression in this patient, as compared with AFF1 expression in all other B-ALL samples in our patient cohort. Conversely, while KMT2A gene expression was not increased, elevated expression of the homeobox gene MEIS1 was observed as is typical of KMT2Ar patients [12] (Figure 3B). It should be noted that, in an in vivo model, KMT2A::AFF1 fusions were incapable of inducing leukemia in isolation, suggesting additional genomic aberrations are required [13].
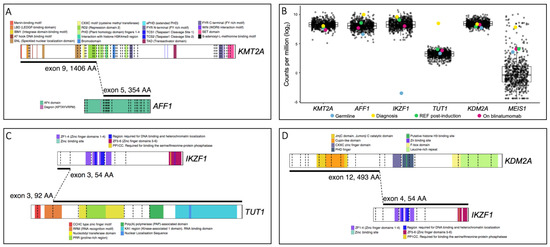
Figure 3.
Schematic representations of the KMT2A::AFF1, IKZF1::TUT1 and KDM2A::IKZF1 gene fusions were created using ProteinPaint [14]. (A) The KMT2A::AFF1 gene fusion retains DNA- and protein-binding domains, nuclear localization domains and the repression domain responsible for HDAC1 and HDAC2 recruitment. The menin- and LEDGF-binding domains are critical for leukemic transformation involving KMT2A, as both menin and LEDGF are essential oncogenic co-factors for KMT2A. The interaction of the three proteins is necessary for leukemic transformation [10,11]. (B) Boxplots of differential expression levels of five genes involved in gene fusions identified in patient CHI_0391. Gene expression levels in germline, diagnosis, refractory post-induction therapy and on-blinatumomab treatment samples from patient CHI_0391 (colored dots) are denoted and compared with samples from our B-ALL patient cohort (black dots; 592 patient samples at various disease timepoints including the 4 highlighted samples from CHI_0391. See Table S1 for cohort characteristics). The data are expressed as log normalized counts per million. (C) The IKZF1::TUT1 gene fusion retains a small portion of IKZF1 but the majority of TUT1, encompassing the nuclear localization sequence and truncating the RNA recognition motif. (D) The KDM2A::IKZF1 gene fusion retains the catalytic domain of KDM2A and all functional domains of IKZF1.
The Terminal Uridylyl Transferase 1 (TUT1) gene has previously been reported as a fusion partner in T-cell lymphoblastic lymphoma patients [15]; however, IKZF1::TUT1 is a novel fusion gene. TUT1 encodes a nucleotidyl transferase enzyme that may play a role in controlling gene expression and cell proliferation; however, its role in oncogenesis remains unclear. The novel IKZF1::TUT1 fusion is the predominant gene fusion in this patient at all disease timepoints (Figure 2A, Table S2). Interestingly, only a small portion of the IKZF1 gene, containing no functional domains, is present. Thus, it is likely that the TUT1 fusion partner is driving the putative leukemic activity of this fusion. Additionally, the diagnosis sample exhibits the highest expression level of the TUT1 gene observed in our patient cohort (Figure 3B). Limited functional data for TUT1 gene fusions exist; however, given that the RNA recognition motif is truncated while the nuclear localization sequence remains (Figure 3C), it is possible that aberrant gene expression occurs as a result of IKZF1::TUT1.
The second novel fusion involving IKZF1, KDM2A::IKZF1, incorporates the catalytic domain of KDM2A and all IKZF1 functional domains (Figure 3D). The encoded Ikaros protein is a transcription factor with key regulatory functions in lymphopoiesis [16]. IKZF1 is a leukemic driver and functions as a tumor suppressor and loss of Ikaros function, either by mutation or deletion, is frequently observed in B-cell ALL [17] as well as other hematological malignancies [18]. Ikaros loss of function alterations are associated with poor prognosis and inferior treatment outcomes [19,20,21]. However, currently no outcome data for IKZF1 gene fusions are available, most likely due to the rarity of these alterations (Table 1). Whether these fusions are driver alterations and contribute to leukemic development also remains to be determined.
Transcriptomic sequencing was performed on mesenchymal stem cells generated from hair follicles, representing a germline sample, as well as sequential samples taken when the patient was refractory following induction therapy and while undergoing blinatumomab therapy prior to relapse (Figure 2A). Interestingly, the KDM2A::IKZF1 and KMT2A::AFF1 gene fusions were no longer detectable by mRNA-Seq following blinatumomab therapy, suggesting the IKZF1::TUT1 gene fusion may be responsible for driving relapse.
Infant KMT2Ar ALL cases normally present with pro-B-cell blasts with the immunophenotype: B220/CD43/19/34/22/TdT/CyCD79a+, CD10/BPI/IgM– [22,23]. However, immunophenotypic analyses of PBMNCs from this infant detected an atypical immunophenotype at diagnosis with a clear population of CD10+ cells (93% CD19/CD34+, 39% CD10/CD19/CD34+) suggesting the presence of two leukemic populations (Figure 1 and Figure S1). Similarly, at both the refractory (13% CD19/CD34+, 0.6% CD10/CD19/CD34+) and on-blinatumomab timepoints (CD19 negative, 2.8% CD10/CD34+), two immunophenotypic populations were present. Comparing these data with the gene fusions identified by transcriptomic sequencing (Figure 2A), it is likely that the CD19- clone present during blinatumomab therapy harbored the IKZF1::TUT1 gene fusion, while the CD10/CD19+ clone harbored the KDM2A::IKZF1 and KMT2A::AFF1 gene fusions. However, this would require confirmatory sequencing of each cell population.

Table 1.
IKZF1 gene fusions previously reported in acute lymphoblastic leukemia.
Table 1.
IKZF1 gene fusions previously reported in acute lymphoblastic leukemia.
| Fusion | Chromosome Abnormality | Disease | Number of Cases | Description | IKZF1 Exon Retention | Predicted Function | Ref |
|---|---|---|---|---|---|---|---|
| DNAH14::IKZF1 | t(1;7)(q42;p12) | B-ALL | 1 | in-frame; exon 36 to exon 4 ZNF2-4 domains (DNA-binding function) and ZNF5-6 domains containing dimerization sites are truncated | exons 1–4 | Similar to the IK6 isoform, loss-of-function allele | [24] |
| ETV6::IKZF1 | t(7;12)(p12;p13) | B-ALL | 1 | out-of-frame; intron 2 to intron 3 No functional IKZF1 domains are present | exons 1–3 | Likely abolishes the function of Ikaros protein | [18] |
| FIGNL1::IKZF1 | t(7;7)(p12;p12) | B-ALL | 1 | out-of-frame; exon 4 to exon 4* Majority of functional IKZF1 domains are retained; ZNF1 domain (DNA-binding function) is truncated | exons 5–8 | Altered transcriptional regulation | [25] |
| IKZF1::CDK2 | t(7;12)(p12;q13) | B-ALL | 1 | out-of-frame; exon 3, no CDK2 breakpoint details provided No functional Ikaros domains are present | exons 1–3 | Likely abolishes the function of Ikaros protein | [26] |
| IKZF1::ETV6 | t(7;12)(p12;p13) | B-ALL | 1 | out-of-frame; intron 3 to intron 2 No functional IKZF1 domains are present | exons 1–3 | Likely abolishes the function of Ikaros protein | [18] |
| IKZF1::FIGNL1 | t(7;7)(p12;p12) | B-ALL | 2 | out-of-frame; intron 3 to 5′ UTR exon 2 and exon 3 to 13691 bp downstream No functional IKZF1 domains are present | exons 1–3 | Likely abolish the function of Ikaros protein | [18,27] |
| IKZF1::NUTM1 | t(7;15)(p12;q14) | B-ALL | 1 | in-frame; exon 7 to exon 2 Some functional IKZF1 domains are retained; ZNF5-6 domains containing dimerization sites are truncated | exons 1–7 | Altered transcriptional regulation | [26] |
| IKZF1::SETD5 | t(3;7)(p25;p12) | B-ALL | 1 # | in-frame; exon 3, no SETD5 breakpoint details provided No functional IKZF1 domains are present | exons 1–3 | Likely abolishes the function of Ikaros protein | [26] |
| IKZF1::TRPV2 | t(7;17)(p12;p11) | B-ALL | 1 | out-of-frame; no exon/intron details provided No functional IKZF1 domains are present | breakpoint not specified | Likely abolishes the function of Ikaros protein | [26] |
| IKZF1::ZEB2 | t(2;7)(q22;p12) | B-ALL | 1 | in-frame; exon 3 to exon 5 No functional IKZF1 domains are present | exons 1–3 | Likely abolishes the function of Ikaros protein | [18] |
| SETD5::IKZF1 | t(3;7)(p25;p12) | B-ALL | 1 # | in-frame; no SETD5 breakpoint details provided, exon 4 All functional IKZF1 domains are retained | exons 4–8 | Altered transcriptional regulation | [26] |
| STIM2::IKZF1 | t(4;7)(p15;p12) | B-ALL | 1 | No fusions details provided | breakpoint not specified | No fusion details provided | [28] |
| IKZF1::ABCA13 | t(7;7)(p12;p12) | T-ALL | 1 | out-of-frame; exon to intron Some functional IKZF1 domains are retained; ZNF4 domain (DNA-binding function) and ZNF5-6 domains containing dimerization sites are truncated | exons 1–5 | Reduced expression of IKZF1; likely abolishes the function of Ikaros protein | [29] |
| IKZF1::NOTCH1 | t(7;9)(p12;q34) | T-ALL | 1 | in-frame; exon to exon Some functional IKZF1 domains are retained; ZNF4 domain (DNA-binding function) and ZNF5-6 domains containing dimerization sites are truncated | exons 1–5 | Altered transcriptional regulation | [30] |
| IKZF1::NOTCH1 | t(7;9)(p12;q34) | T-ALL | 1 $ | out-of-frame; intron to intron Some functional IKZF1 domains are retained; ZNF4 domain (DNA-binding function) and ZNF5-6 domains containing dimerization sites are truncated | exons 1–5 | Altered transcriptional regulation | [27] |
| NOTCH1::IKZF1 | t(7;9)(p12;q34) | T-ALL | 1 $ | out-of-frame; exon to intron Some functional IKZF1 domains are retained; ZNF1-4 domains containing dimerization sites are truncated | exons 6–8 | Altered transcriptional regulation | [27] |
Note 1: Retained IKZF1 exons refers to IKZF1 transcript variant 1, NM_006060 except for (*), which relates to alternate transcript variant 15, NM_001291845. Note 2: # IKZF1::SETD5 and SETD5::IKZF1 fusions were observed in the same patient. Note 3: $ IKZF1::NOTCH1 and NOTCH1::IKZF1 fusions were observed in the same patient.
4. Discussion
Here, we describe a rare case of congenital KMT2Ar ALL presenting with co-occurring IKZF1 gene fusions and a predictably aggressive disease trajectory. We report for the first time the novel IKZF1::TUT1 and KDM2A::IKZF1 gene fusions. Rearrangements involving KMT2A are commonly retained in relapsed infant ALL [5,31,32]; however, in this case, the KMT2A::AFF1 gene fusion did not appear to be the lesion driving leukemic relapse. Instead, our data suggest that relapse was driven by IKZF1::TUT1. This gene fusion remained in all samples investigated, including the on-blinatumomab therapy sample taken immediately prior to relapse. Conversely, the KMT2A::AFF1 gene fusion was only detected in the diagnosis and refractory post-induction samples, highlighting a key role for IKZF1::TUT1 in disease pathogenesis. Intriguingly, both IKZF1 gene fusions are predicted to be out-of-frame (Table S2); however, our data demonstrate the IKZF1 gene is still expressed (Figure 3B). This is not unprecedented, and it has previously been observed that out-of-frame fusions can cause transcriptional activation/repression of genes involved in the fusions leading to increases or decreases of their expression and the associated functional outcomes [29].
Ikaros is a lymphoid transcription factor with a tumor-suppressive function. Alterations that knock out the Ikaros function, such as the gene fusions described here, would presumably also affect Ikaros target genes including signal transducers (c-kit, Flt3, Il7r), pre-B-cell receptor signaling proteins (Syk) and cell cycle regulators (Cdkn2a, Cdkn1a) [33]. Indeed, altered Flt3 [34] and Syk [35] expression has been reported in KMT2Ar ALL. Studies have demonstrated Flt3 inhibitors are active against KMT2Ar disease in vivo [36] and, when administered in combination with various chemotherapeutics including some of those used here (dexamethasone, cytarabine, asparaginase), effectively kill KMT2Ar cells in vitro [37,38]. More recently, a Children’s Oncology Group trial has demonstrated the benefit of Flt3 inhibitor lestaurtinib in combination with chemotherapy (Interfant 99-based induction regimen) for treating infants with KMT2Ar ALL [39]. Similarly, the combination of vincristine and the Syk inhibitor entospletinib demonstrated enhanced efficacy in in vivo models of infant KMT2Ar ALL compared with either agent alone [40]. However, entospletinib as a treatment for ALL has yet to enter clinical trials. A retrospective case study of 11 infants with KMT2Ar ALL demonstrated the efficacy of blinatumomab in patients with relapsed/refractory disease [41], and pre-clinical efficacy was observed in recent in vivo models assessing azacitidine in combination with venetoclax [42]. However, we observed poor response to the Interfant-06 induction and Children’s Oncology Group consolidation protocols (AALL15P1), which comprise various chemotherapy agents, including those detailed above (Figure 1), and while azacitidine was well tolerated, the patient soon became resistant. The immunotherapies blinatumomab and inotuzomab [43] as well as the FLAG-IDA relapse regimen also failed. Case reports such as this highlight the urgent need for new targeted therapies to improve outcome in KMT2Ar infant ALL. We also demonstrate the importance of gene sequencing to comprehensively dissect the underlying genomic complexity of a disease like ALL and identify co-occurring alterations that may impact treatment outcomes.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes14020264/s1, Table S1: Key characteristics of the B-ALL patient cohort used in the gene expression analyses; Table S2: Complete range of gene fusions identified in patient CHI_0391 by transcriptomic sequencing; Figure S1: Immunophenotyping analyses of diagnosis, refractory and on-blinatumomab samples; Figure S2: Analysis of deletions in key B-ALL genes in CHI_0391 identified by multiplex ligand-dependent probe amplification (MLPA).
Author Contributions
Manuscript conceptualization, L.N.E. and D.L.W.; formal analysis, L.N.E., J.A.R., J.B., S.L.H., B.J.M. and D.L.W.; clinical data and interpretation, M.P.O., S.J., D.T.Y., T.R. and B.S.; experimental design and completion, C.E.J.D.; writing—original draft preparation, L.N.E., M.P.O. and S.J.; figure preparation, L.N.E., J.A.R. and C.E.J.D.; writing—review and editing, L.N.E., M.P.O., S.J., D.T.Y. and D.L.W. All authors have read and agreed to the published version of the manuscript.
Funding
L.N.E. receives fellowship funding from the Peter Nelson Leukaemia Research Fellowship Fund, administered by the Cancer Council South Australia (grant number 022212). S.L.H. receives fellowship funding from The Kids’ Cancer Project (grant number 022214). D.T.Y. receives fellowship funding from the National Health and Medical Research Council (NHMRC; grant number 1146253). D.L.W. receives fellowship funding from the NHMRC (grant number 1160833) and the Cancer Council Beat Cancer Project (grant number 022213).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Royal Adelaide Hospital Human Research Ethics Committee (HREC/15/RAH/54; RAH Protocol: 150212; approved 12 February 2015).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in the study are deposited in the European Genome Phenome Archive.
Acknowledgments
Authors would like to acknowledge the South Australian Genomics Centre (SAGC), Adelaide, South Australia for performing the transcriptomic sequencing analyses and the Genetics and Molecular Pathology at SA Pathology, Adelaide, South Australia for performing the Cytogenetics and FISH.
Conflicts of Interest
L.N.E., J.A.R., J.B., M.P.O., S.J., C.E.J.D., S.L.H., B.J.M., T.R. and B.S. have no conflicts of interest to declare. D.L.W. receives honoraria and research funding from Bristol-Myers Squibb and honoraria from Amgen. D.T.Y. receives honoraria and research funding from Bristol-Myers Squibb and Novartis and honoraria from Amgen and Pfizer. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Forgione, M.O.; McClure, B.J.; Eadie, L.N.; Yeung, D.T.; White, D.L. KMT2A rearranged acute lymphoblastic leukaemia: Unravelling the genomic complexity and heterogeneity of this high-risk disease. Cancer Lett. 2020, 469, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Tauchi, H.; Tomizawa, D.; Eguchi, M.; Eguchi-Ishimae, M.; Koh, K.; Hirayama, M.; Miyamura, N.; Kinukawa, N.; Hayashi, Y.; Horibe, K.; et al. Clinical features and outcome of MLL gene rearranged acute lymphoblastic leukemia in infants with additional chromosomal abnormalities other than 11q23 translocation. Leuk. Res. 2008, 32, 1523–1529. [Google Scholar] [CrossRef]
- Sun, L.; Heerema, N.; Crotty, L.; Wu, X.; Navara, C.; Vassilev, A.; Sensel, M.; Reaman, G.H.; Uckun, F.M. Expression of dominant-negative and mutant isoforms of the antileukemic transcription factor Ikaros in infant acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 1999, 96, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.; Lorenzo, P.D.; Ancliffe, P.; Aversa, L.A.; Brethon, B.; Biondi, A.; Campbell, M.; Escherich, G.; Ferster, A.; Gardner, R.A.; et al. Outcome of Infants Younger Than 1 Year With Acute Lymphoblastic Leukemia Treated with the Interfant-06 Protocol: Results From an International Phase III Randomized Study. J. Clin. Oncol. 2019, 37, 2246–2256. [Google Scholar] [CrossRef] [PubMed]
- Guest, E.; Kairalla, J.; Devidas, M.; Hibbitts, E.; Carroll, A.J.; Heerema, N.A.; Kubaney, H.; August, A.; Pauly, M.; Wechsler, D.; et al. A Pilot Study of Azacitidine As Epigenetic Priming for Chemotherapy in Infants Less Than 1 Year of Age with KMT2A-Rearranged Acute Lymphoblastic Leukemia (ALL); Results from the Children’s Oncology Group (COG) Trial AALL15P1. Blood 2022, 140, 3256–3257. [Google Scholar] [CrossRef]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2017, 32, 273. [Google Scholar] [CrossRef]
- Robinson, M.A. Linkage Disequilibrium. In Encyclopedia of Immunology, 2nd ed.; Delves, P.J., Ed.; Elsevier: Oxford, UK, 1998; pp. 1586–1588. [Google Scholar]
- Murai, M.J.; Pollock, J.; He, S.; Miao, H.; Purohit, T.; Yokom, A.; Hess, J.L.; Muntean, A.G.; Grembecka, J.; Cierpicki, T. The same site on the integrase-binding domain of lens epithelium-derived growth factor is a therapeutic target for MLL leukemia and HIV. Blood 2014, 124, 3730–3737. [Google Scholar] [CrossRef]
- Yokoyama, A.; Cleary, M.L. Menin Critically Links MLL Proteins with LEDGF on Cancer-Associated Target Genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef]
- Ferrando, A.A.; Armstrong, S.A.; Neuberg, D.S.; Sallan, S.E.; Silverman, L.B.; Korsmeyer, S.J.; Look, A.T. Gene expression signatures in MLL-rearranged T-lineage and B-precursor acute leukemias: Dominance of HOX dysregulation. Blood 2003, 102, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Montes, R.; Ayllón, V.; Gutierrez-Aranda, I.; Prat, I.; Hernández-Lamas, M.C.; Ponce, L.; Bresolin, S.; te Kronnie, G.; Greaves, M.; Bueno, C.; et al. Enforced expression of MLL-AF4 fusion in cord blood CD34+ cells enhances the hematopoietic repopulating cell function and clonogenic potential but is not sufficient to initiate leukemia. Blood 2011, 117, 4746–4758. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Edmonson, M.N.; Wilkinson, M.R.; Patel, A.; Wu, G.; Liu, Y.; Li, Y.; Zhang, Z.; Rusch, M.C.; Parker, M.; et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet. 2016, 48, 4–6. [Google Scholar] [CrossRef] [PubMed]
- López-Nieva, P.; Fernández-Navarro, P.; Graña-Castro, O.; Andrés-León, E.; Santos, J.; Villa-Morales, M.; Cobos-Fernández, M.Á.; González-Sánchez, L.; Malumbres, M.; Salazar-Roa, M.; et al. Detection of novel fusion-transcripts by RNA-Seq in T-cell lymphoblastic lymphoma. Sci. Rep. 2019, 9, 5179. [Google Scholar] [CrossRef]
- Georgopoulos, K.; Bigby, M.; Wang, J.-H.; Molnar, A.; Wu, P.; Winandy, S.; Sharpe, A. The ikaros gene is required for the development of all lymphoid lineages. Cell 1994, 79, 143–156. [Google Scholar] [CrossRef]
- Marke, R.; van Leeuwen, F.N.; Scheijen, B. The many faces of IKZF1 in B-cell precursor acute lymphoblastic leukemia. Haematologica 2018, 103, 565–574. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion of IKZF1 and Prognosis in Acute Lymphoblastic Leukemia. New Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef]
- Kuiper, R.P.; Waanders, E.; van der Velden, V.H.J.; van Reijmersdal, S.V.; Venkatachalam, R.; Scheijen, B.; Sonneveld, E.; van Dongen, J.J.M.; Veerman, A.J.P.; van Leeuwen, F.N.; et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia 2010, 24, 1258–1264. [Google Scholar] [CrossRef]
- Waanders, E.; Van Der Velden, V.H.J.; Van Der Schoot, C.E.; Van Leeuwen, F.N.; Van Reijmersdal, S.V.; De Haas, V.; Veerman, A.J.; Van Kessel, A.G.; Hoogerbrugge, P.M.; Kuiper, R.P.; et al. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia 2011, 25, 254–258. [Google Scholar] [CrossRef]
- Chiaretti, S.; Zini, G.; Bassan, R. Diagnosis and subclassification of acute lymphoblastic leukemia. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014073. [Google Scholar] [CrossRef] [PubMed]
- Patton, D.T.; Plumb, A.W.; Abraham, N. The Survival and Differentiation of Pro-B and Pre-B Cells in the Bone Marrow Is Dependent on IL-7Rα Tyr449. J. Immunol. 2014, 193, 3446–3455. [Google Scholar] [CrossRef] [PubMed]
- Mata-Rocha, M.; Rangel-López, A.; Jiménez-Hernández, E.; Morales-Castillo, B.A.; González-Torres, C.; Gaytan-Cervantes, J.; Álvarez-Olmos, E.; Núñez-Enríquez, J.C.; Fajardo-Gutiérrez, A.; Martín-Trejo, J.A.; et al. Identification and Characterization of Novel Fusion Genes with Potential Clinical Applications in Mexican Children with Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2019, 20, 2394. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Zhang, H.; Kham, S.K.; Liu, S.; Jiang, C.; Zhao, X.; Lu, Y.; Goodings, C.; Lin, T.N.; Zhang, R.; et al. Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res. 2017, 27, 185–195. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 11790. [Google Scholar] [CrossRef]
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.; Roberts, K.; Li, Y.; Liu, Y.; Harvey, R.C.; McCastlain, K.; Reshmi, S.C.; Payne-Turner, D.; Iacobucci, I.; et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 13331. [Google Scholar] [CrossRef]
- Atak, Z.K.; Gianfelici, V.; Hulselmans, G.; De Keersmaecker, K.; Devasia, A.G.; Geerdens, E.; Mentens, N.; Chiaretti, S.; Durinck, K.; Uyttebroeck, A.; et al. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T-cell acute lymphoblastic leukemia. PLoS Genet. 2013, 9, e1003997. [Google Scholar] [CrossRef]
- Chen, B.; Jiang, L.; Zhong, M.-L.; Li, J.-F.; Li, B.-S.; Peng, L.-J.; Dai, Y.-T.; Cui, B.-W.; Yan, T.-Q.; Zhang, W.-N.; et al. Identification of fusion genes and characterization of transcriptome features in T-cell acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2018, 115, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Agraz-Doblas, A.; Bueno, C.; Bashford-Rogers, R.; Roy, A.; Schneider, P.; Bardini, M.; Ballerini, P.; Cazzaniga, G.; Moreno, T.; Revilla, C.; et al. Unraveling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic leukemia using genome-wide analysis. Haematologica 2019, 104, 1176–1188. [Google Scholar] [CrossRef]
- Trentin, L.; Bresolin, S.; Giarin, E.; Bardini, M.; Serafin, V.; Accordi, B.; Fais, F.; Tenca, C.; De Lorenzo, P.; Valsecchi, M.G.; et al. Deciphering KRAS and NRAS mutated clone dynamics in MLL-AF4 paediatric leukaemia by ultra deep sequencing analysis. Sci. Rep. 2016, 6, 34449. [Google Scholar] [CrossRef] [PubMed]
- Ferreirós-Vidal, I.; Carroll, T.; Taylor, B.; Terry, A.; Liang, Z.; Bruno, L.; Dharmalingam, G.; Khadayate, S.; Cobb, B.S.; Smale, S.T.; et al. Genome-wide identification of Ikaros targets elucidates its contribution to mouse B-cell lineage specification and pre-B–cell differentiation. Blood 2013, 121, 1769–1782. [Google Scholar] [CrossRef]
- Chillón, M.C.; Gómez-Casares, M.T.; López-Jorge, C.E.; Rodriguez-Medina, C.; Molines, A.; Sarasquete, M.E.; Alcoceba, M.; Miguel, J.D.G.S.; Bueno, C.; Montes, R.; et al. Prognostic significance of FLT3 mutational status and expression levels in MLL-AF4+ and MLL-germline acute lymphoblastic leukemia. Leukemia 2012, 26, 2360–2366. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Qazi, S. Tyrosine kinases in KMT2A/MLL-rearranged acute leukemias as potential therapeutic targets to overcome cancer drug resistance. Cancer Drug Resist. 2022, 5, 902–916. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Kung, A.L.; Mabon, M.E.; Silverman, L.B.; Stam, R.W.; Den Boer, M.L.; Pieters, R.; Kersey, J.H.; Sallan, S.E.; Fletcher, J.A.; et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell 2003, 3, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Levis, M.; McIntyre, E.; Griesemer, M.; Small, D. Combinations of the FLT3 inhibitor CEP-701 and chemotherapy synergistically kill infant and childhood MLL-rearranged ALL cells in a sequence-dependent manner. Leukemia 2006, 20, 1368–1376. [Google Scholar] [CrossRef]
- Stam, R.W.; den Boer, M.L.; Schneider, P.; Nollau, P.; Horstmann, M.; Beverloo, H.B.; van der Voort, E.; Valsecchi, M.G.; de Lorenzo, P.; Sallan, S.E.; et al. Targeting FLT3 in primary MLL-gene-rearranged infant acute lymphoblastic leukemia. Blood 2005, 106, 2484–2490. [Google Scholar] [CrossRef]
- Brown, P.A.; Kairalla, J.A.; Hilden, J.M.; Dreyer, Z.E.; Carroll, A.J.; Heerema, N.A.; Wang, C.; Devidas, M.; Gore, L.; Salzer, W.L.; et al. FLT3 inhibitor lestaurtinib plus chemotherapy for newly diagnosed KMT2A-rearranged infant acute lymphoblastic leukemia: Children’s Oncology Group trial AALL0631. Leukemia 2021, 35, 1279–1290. [Google Scholar] [CrossRef]
- Loftus, J.P.; Yahiaoui, A.; Brown, P.A.; Niswander, L.M.; Bagashev, A.; Wang, M.; Schauf, A.; Tannheimer, S.; Tasian, S.K. Combinatorial efficacy of entospletinib and chemotherapy in patient-derived xenograft models of infant acute lymphoblastic leukemia. Haematologica 2020, 106, 1067–1078. [Google Scholar] [CrossRef]
- Clesham, K.; Rao, V.; Bartram, J.; Ancliff, P.; Ghorashian, S.; O’Connor, D.; Pavasovic, V.; Rao, A.; Samarasinghe, S.; Cummins, M.; et al. Blinatumomab for infant acute lymphoblastic leukemia. Blood 2020, 135, 1501–1504. [Google Scholar] [CrossRef]
- Cheung, L.C.; Aya-Bonilla, C.; Cruickshank, M.N.; Chiu, S.K.; Kuek, V.; Anderson, D.; Chua, G.A.; Singh, S.; Oommen, J.; Ferrari, E.; et al. Preclinical efficacy of azacitidine and venetoclax for infant KMT2A-rearranged acute lymphoblastic leukemia reveals a new therapeutic strategy. Leukemia 2022, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, H.; Karsli, U.; Schoenherr, C.; Battmer, K.; Erschow, S.; Talbot, S.R.; Steinemann, D.; Heuser, M.; Heidenreich, O.; Hilfiker-Kleiner, D.; et al. Venetoclax and dexamethasone synergize with inotuzumab ozogamicin–induced DNA damage signaling in B-lineage ALL. Blood 2021, 137, 2657–2661. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).