
Classification of Acute Myeloid Leukemia by Cell-Free DNA 5-Hydroxymethylcytosine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. DNA Extraction and 5hmC Sequencing
2.3. Analysis of Genome-Wide 5hmC Distribution
2.4. Statistical Analyses
3. Results
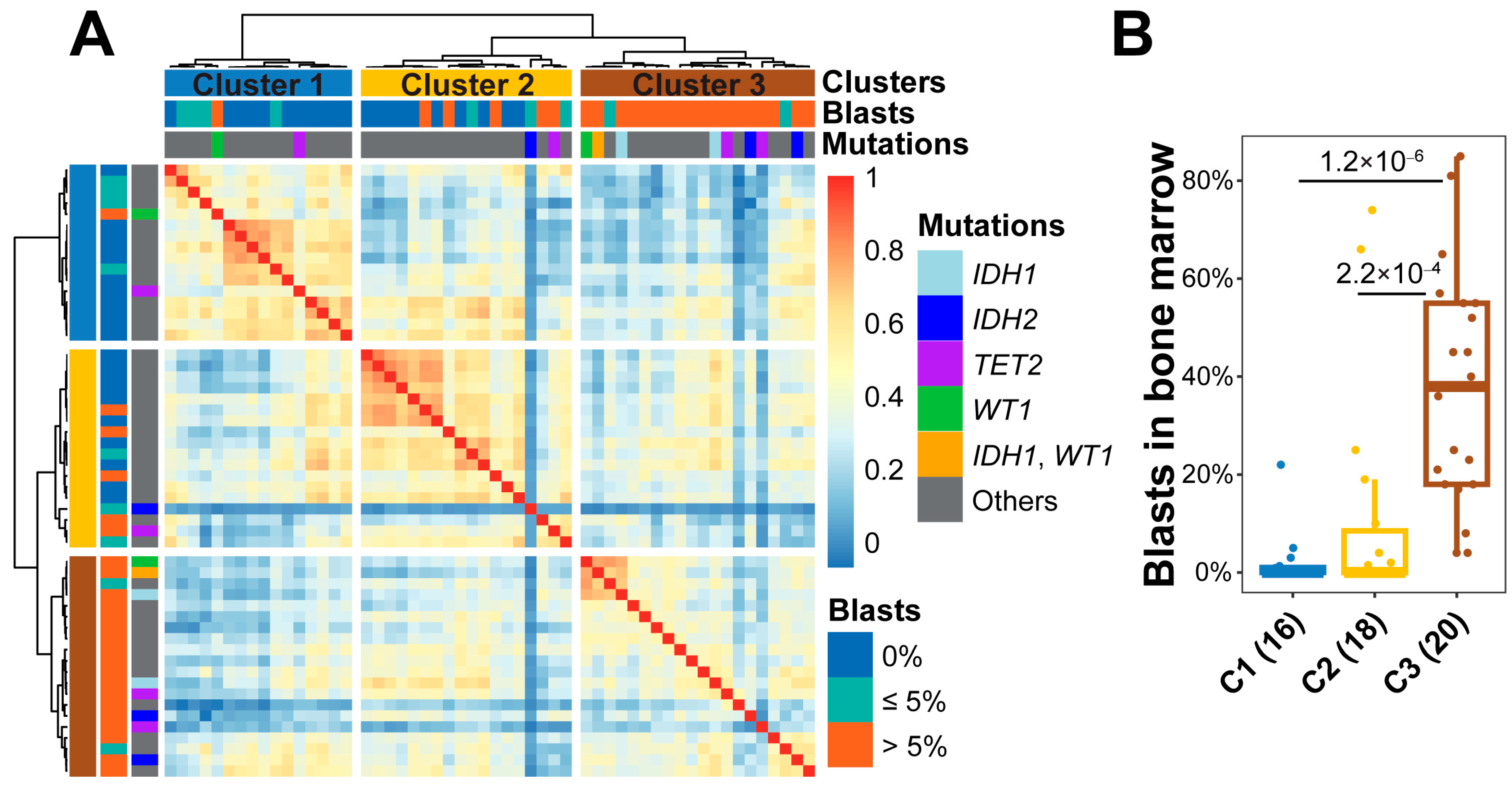
3.1. Classification of AML Samples Using Genome-Wide 5hmC Distribution
3.2. Characteristics of cfDNA in the Clusters of AML Patients
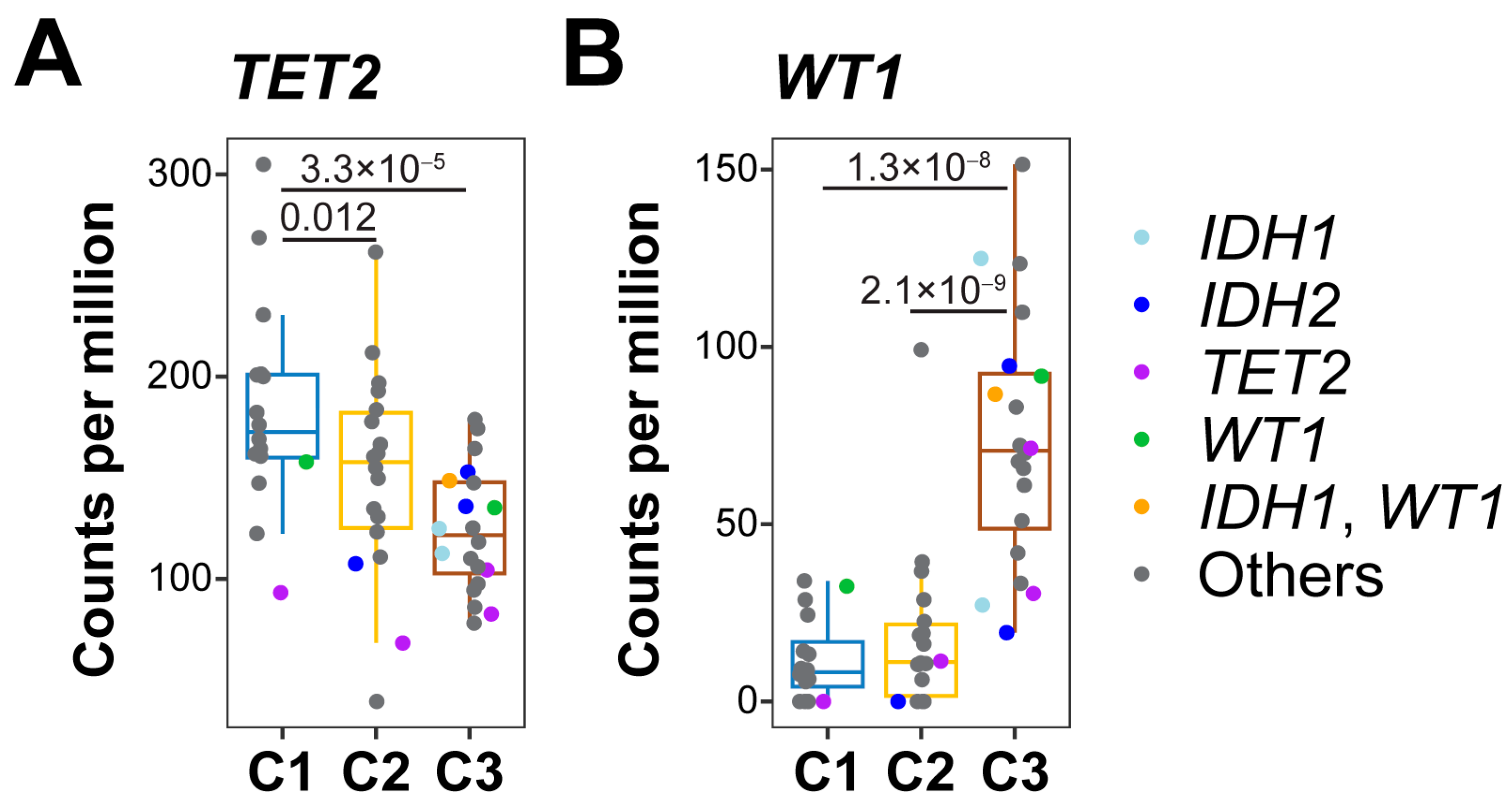
3.3. 5-hmC Levels in DNA Demethylation Genes
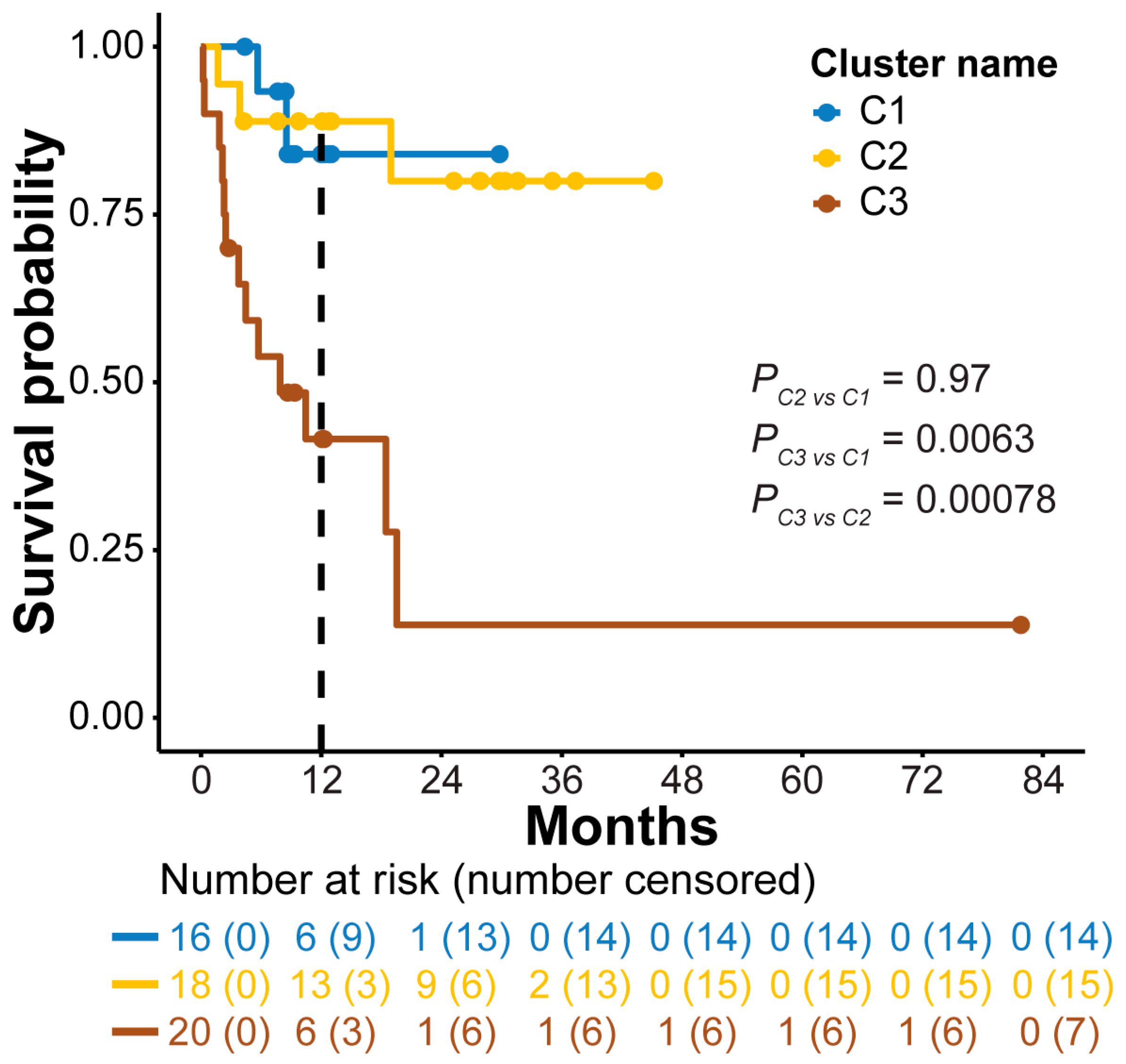
3.4. Association of 5hmC Clusters with OS in AML Patients
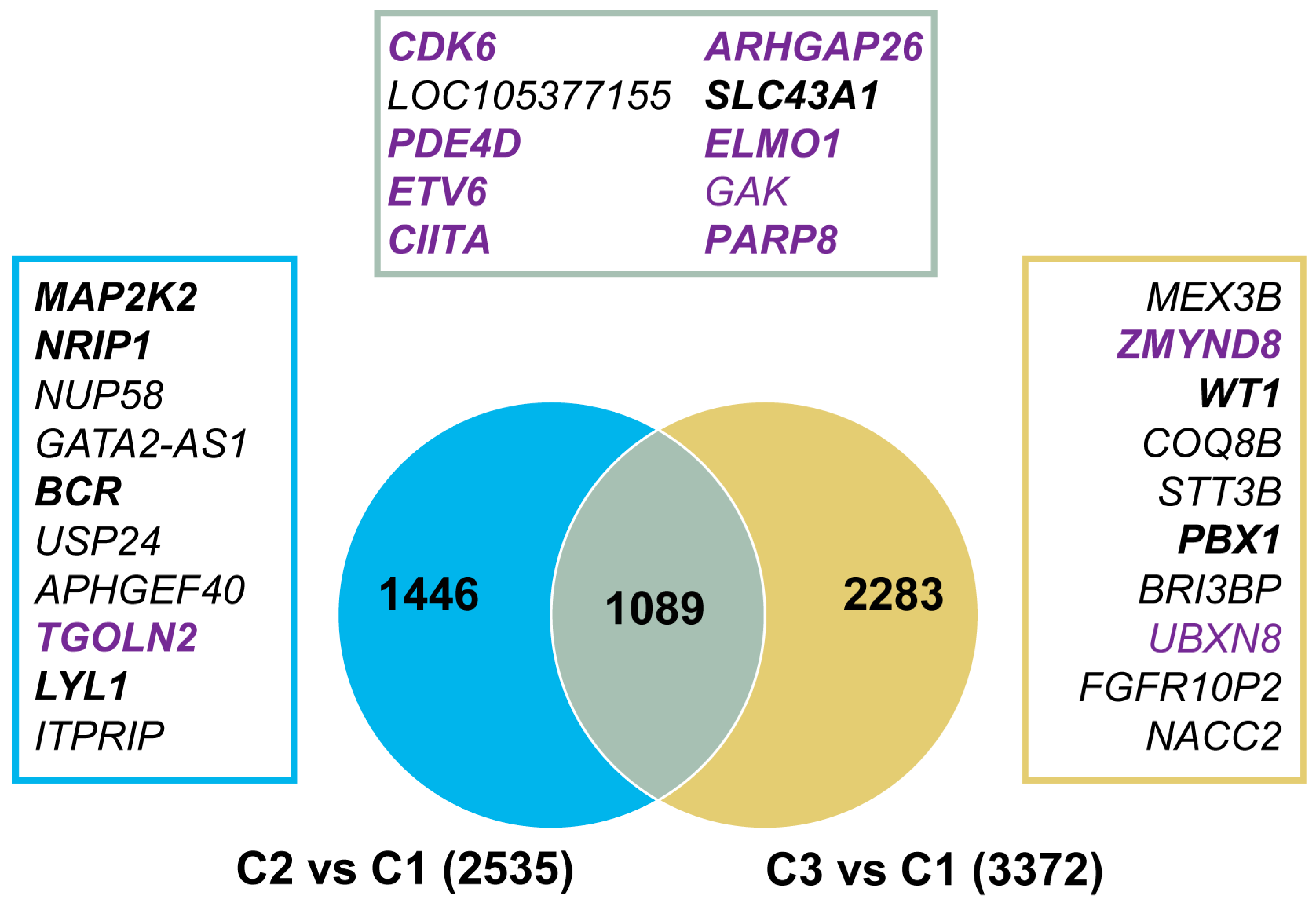
3.5. Genomic Regions with Aberrant 5hmC Levels in AML
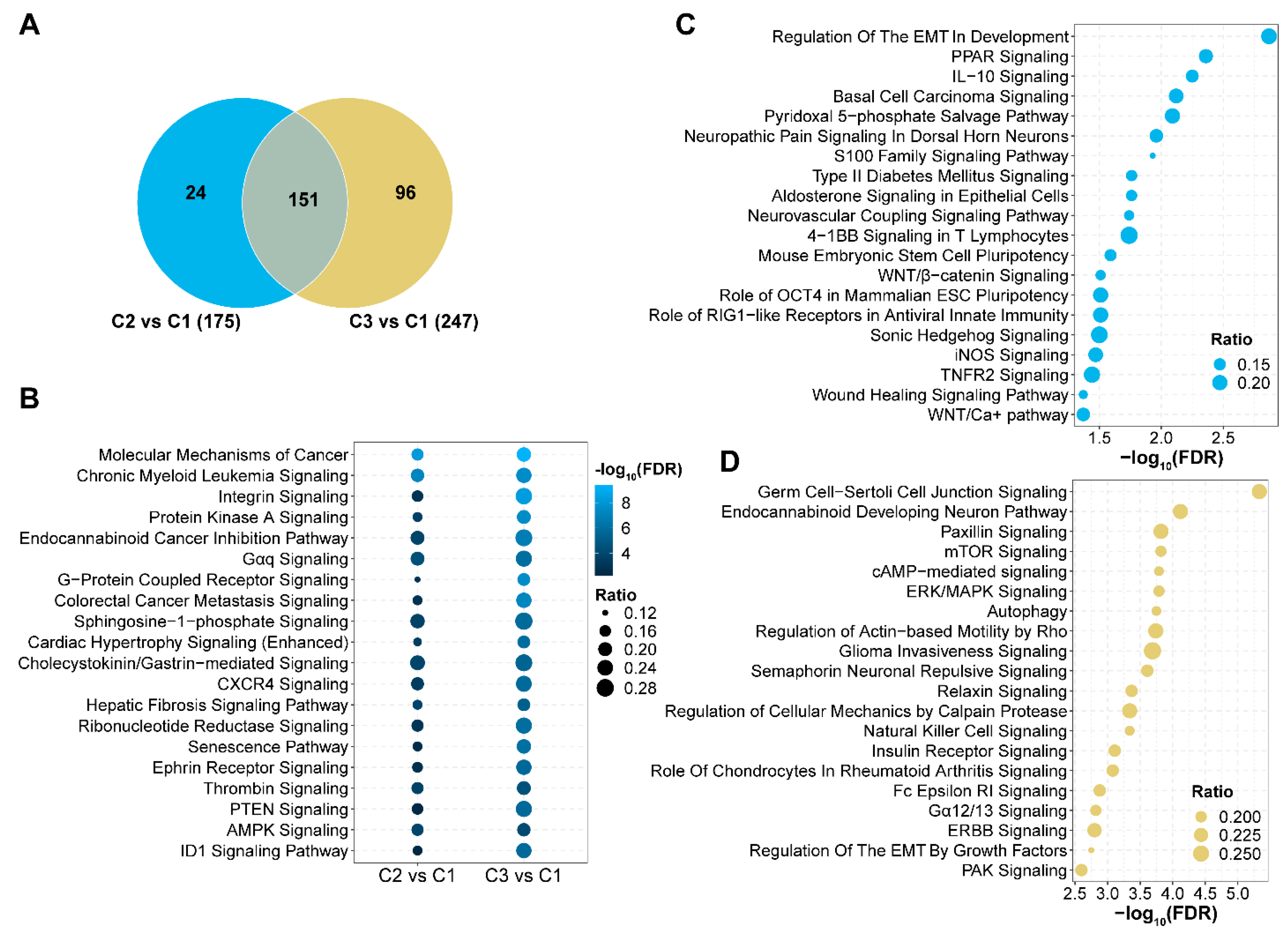
3.6. Signaling Pathways with Aberrant 5hmC Levels in AML
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Zeng, C.; Stroup, E.K.; Zhang, Z.; Chiu, B.C.; Zhang, W. Towards precision medicine: Advances in 5-hydroxymethylcytosine cancer biomarker discovery in liquid biopsy. Cancer Commun. 2019, 39, 12. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhou, Y.; Chen, L.; Bissessur, A.S.; Chen, J.; Mao, M.; Ju, S.; Chen, L.; Chen, C.; Li, Z.; et al. Deoxyribonucleic Acid 5-Hydroxymethylation in Cell-Free Deoxyribonucleic Acid, a Novel Cancer Biomarker in the Era of Precision Medicine. Front. Cell Dev. Biol. 2021, 9, 744990. [Google Scholar] [CrossRef] [PubMed]
- Kroeze, L.I.; Aslanyan, M.G.; van Rooij, A.; Koorenhof-Scheele, T.N.; Massop, M.; Carell, T.; Boezeman, J.B.; Marie, J.P.; Halkes, C.J.; de Witte, T.; et al. Characterization of acute myeloid leukemia based on levels of global hydroxymethylation. Blood 2014, 124, 1110–1118. [Google Scholar] [CrossRef]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Konstandin, N.; Bultmann, S.; Szwagierczak, A.; Dufour, A.; Ksienzyk, B.; Schneider, F.; Herold, T.; Mulaw, M.; Kakadia, P.M.; Schneider, S.; et al. Genomic 5-hydroxymethylcytosine levels correlate with TET2 mutations and a distinct global gene expression pattern in secondary acute myeloid leukemia. Leukemia 2011, 25, 1649–1652. [Google Scholar] [CrossRef]
- Bowman, R.L.; Levine, R.L. TET2 in Normal and Malignant Hematopoiesis. Cold Spring Harb Perspect Med. 2017, 7, a026518. [Google Scholar] [CrossRef]
- Chowdhury, B.; McGovern, A.; Cui, Y.; Choudhury, S.R.; Cho, I.H.; Cooper, B.; Chevassut, T.; Lossie, A.C.; Irudayaraj, J. The hypomethylating agent Decitabine causes a paradoxical increase in 5-hydroxymethylcytosine in human leukemia cells. Sci Rep. 2015, 5, 9281. [Google Scholar] [CrossRef]
- Gerecke, C.; Schumacher, F.; Edlich, A.; Wetzel, A.; Yealland, G.; Neubert, L.K.; Scholtka, B.; Homann, T.; Kleuser, B. Vitamin C promotes decitabine or azacytidine induced DNA hydroxymethylation and subsequent reactivation of the epigenetically silenced tumour suppressor CDKN1A in colon cancer cells. Oncotarget. 2018, 9, 32822–32840. [Google Scholar] [CrossRef]
- Vachhani, P.; Al Yacoub, R.; Miller, A.; Zhang, F.; Cronin, T.L.; Ontiveros, E.P.; Thompson, J.E.; Griffiths, E.A.; Wang, E.S. Intensive chemotherapy vs. hypomethylating agents in older adults with newly diagnosed high-risk acute myeloid leukemia: A single center experience. Leuk Res. 2018, 75, 29–35. [Google Scholar] [CrossRef]
- Pan, D.; Rampal, R.; Mascarenhas, J. Clinical developments in epigenetic-directed therapies in acute myeloid leukemia. Blood Adv. 2020, 4, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Lövkvist, C.; Dodd, I.B.; Sneppen, K.; Haerter, J.O. DNA methylation in human epigenomes depends on local topology of CpG sites. Nucleic Acids Res. 2016, 44, 5123–5132. [Google Scholar] [CrossRef] [PubMed]
- Vonk, C.M.; Al Hinai, A.S.; Hanekamp, D.; Valk, P.J.M. Molecular Minimal Residual Disease Detection in Acute Myeloid Leukemia. Cancers. 2021, 13, 5431. [Google Scholar] [CrossRef] [PubMed]
- Giacopelli, B.; Wang, M.; Cleary, A.; Wu, Y.Z.; Schultz, A.R.; Schmutz, M.; Blachly, J.S.; Eisfeld, A.K.; Mundy-Bosse, B.; Vosberg, S.; et al. DNA methylation epitypes highlight underlying developmental and disease pathways in acute myeloid leukemia. Genome Res. 2021, 31, 747–761. [Google Scholar] [CrossRef]
- Li, Y.; Xu, Q.; Lv, N.; Wang, L.; Zhao, H.; Wang, X.; Guo, J.; Chen, C.; Li, Y.; Yu, L. Clinical implications of genome-wide DNA methylation studies in acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 41. [Google Scholar] [CrossRef]
- Krali, O.; Palle, J.; Backlin, C.L.; Abrahamsson, J.; Noren-Nystrom, U.; Hasle, H.; Jahnukainen, K.; Jonsson, O.G.; Hovland, R.; Lausen, B.; et al. DNA Methylation Signatures Predict Cytogenetic Subtype and Outcome in Pediatric Acute Myeloid Leukemia (AML). Genes 2021, 12, 895. [Google Scholar] [CrossRef]
- Chowdhury, B.; Cho, I.H.; Irudayaraj, J. Technical advances in global DNA methylation analysis in human cancers. J Biol Eng. 2017, 11, 1–12. [Google Scholar] [CrossRef]
- Bachman, M.; Uribe-Lewis, S.; Yang, X.; Williams, M.; Murrell, A.; Balasubramanian, S. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem. 2014, 6, 1049–1055. [Google Scholar] [CrossRef]
- He, B.; Zhang, C.; Zhang, X.; Fan, Y.; Zeng, H.; Liu, J.; Meng, H.; Bai, D.; Peng, J.; Zhang, Q.; et al. Tissue-specific 5-hydroxymethylcytosine landscape of the human genome. Nat. Commun. 2021, 12, 4249. [Google Scholar] [CrossRef]
- Han, D.; Lu, X.; Shih, A.H.; Nie, J.; You, Q.; Xu, M.M.; Melnick, A.M.; Levine, R.L.; He, C. A Highly Sensitive and Robust Method for Genome-wide 5hmC Profiling of Rare Cell Populations. Mol. Cell 2016, 63, 711–719. [Google Scholar] [CrossRef]
- Shao, J.; Wang, S.; West-Szymanski, D.; Karpus, J.; Shah, S.; Ganguly, S.; Smith, J.; Zu, Y.; He, C.; Li, Z. Cell-free DNA 5-hydroxymethylcytosine is an emerging marker of acute myeloid leukemia. Sci. Rep. 2022, 12, 12410. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Bernicker, E.; He, C.; Li, Z. Cell-free DNA 5-hydroxymethylcytosine as a marker for common cancer detection. Clin. Transl. Discov. 2022, 2, e136. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Wang, H.; Fan, Z.; Shliaha, P.V.; Miele, M.; Hendrickson, R.C.; Jiang, X.; Helin, K. H3K4me3 regulates RNA polymerase II promoter-proximal pause-release. Nature 2023, 615, 339–348. [Google Scholar] [CrossRef]
- Venney, D.; Mohd-Sarip, A.; Mills, K.I. The Impact of Epigenetic Modifications in Myeloid Malignancies. Int. J. Mol. Sci. 2021, 22, 5013. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, G.; Yi, Y.; Xiao, L.; Pei, M.; Liu, S.; Luo, Y.; Zhong, H.; Xu, Y.; Zheng, W.; et al. Decreased 5-hydroxymethylcytosine levels are associated with TET2 mutation and unfavorable overall survival in myelodysplastic syndromes. Leuk. Lymphoma 2013, 54, 2466–2473. [Google Scholar] [CrossRef]
- Magotra, M.; Sakhdari, A.; Lee, P.J.; Tomaszewicz, K.; Dresser, K.; Hutchinson, L.M.; Woda, B.A.; Chen, B.J. Immunohistochemical loss of 5-hydroxymethylcytosine expression in acute myeloid leukaemia: Relationship to somatic gene mutations affecting epigenetic pathways. Histopathology 2016, 69, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.S.; Kim, H.J.; Kim, Y.K.; Lee, S.S.; Ahn, S.Y.; Jung, S.H.; Yang, D.H.; Lee, J.J.; Park, H.J.; Choi, S.H.; et al. 5-Hydroxymethylcytosine correlates with epigenetic regulatory mutations, but may not have prognostic value in predicting survival in normal karyotype acute myeloid leukemia. Oncotarget 2017, 8, 8305–8314. [Google Scholar] [CrossRef]
- Lio, C.J.; Yuita, H.; Rao, A. Dysregulation of the TET family of epigenetic regulators in lymphoid and myeloid malignancies. Blood 2019, 134, 1487–1497. [Google Scholar] [CrossRef]
- Pasca, S.; Jurj, A.; Zdrenghea, M.; Tomuleasa, C. The Potential Equivalents of TET2 Mutations. Cancers 2021, 13, 1499. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Abdel-Wahab, O. Translating recent advances in the pathogenesis of acute myeloid leukemia to the clinic. Genes Dev. 2022, 36, 259–277. [Google Scholar] [CrossRef]
- Rampal, R.; Alkalin, A.; Madzo, J.; Vasanthakumar, A.; Pronier, E.; Patel, J.; Li, Y.; Ahn, J.; Abdel-Wahab, O.; Shih, A.; et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014, 9, 1841–1855. [Google Scholar] [CrossRef]
- Rampal, R.; Figueroa, M.E. Wilms tumor 1 mutations in the pathogenesis of acute myeloid leukemia. Haematologica 2016, 101, 672–679. [Google Scholar] [CrossRef]
- Nebenfuehr, S.; Kollmann, K.; Sexl, V. The role of CDK6 in cancer. Int. J. Cancer 2020, 147, 2988–2995. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Budinich, K.A.; Huang, H.; Ren, D.; Lu, B.; Zhang, Z.; Chen, Q.; Zhou, Y.; Huang, Y.H.; Alikarami, F.; et al. ZMYND8-regulated IRF8 transcription axis is an acute myeloid leukemia dependency. Mol. Cell 2021, 81, 3604–3622.e3610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, A.; Zhu, S.; Min, L.; Liu, S.; Li, P.; Zhang, S. The role of GTPase-activating protein ARHGAP26 in human cancers. Mol. Cell. Biochem. 2022, 477, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Freisleben, F.; Behrmann, L.; Thaden, V.; Muschhammer, J.; Bokemeyer, C.; Fiedler, W.; Wellbrock, J. Downregulation of GLI3 Expression Mediates Chemotherapy Resistance in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 5084. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, M.; Yang, K.; Chi, T.; Liao, Z.; Wei, P. PPAR-alpha Modulators as Current and Potential Cancer Treatments. Front. Oncol. 2021, 11, 599995. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, J.; Shah, S.; Ganguly, S.; Zu, Y.; He, C.; Li, Z. Classification of Acute Myeloid Leukemia by Cell-Free DNA 5-Hydroxymethylcytosine. Genes 2023, 14, 1180. https://doi.org/10.3390/genes14061180
Shao J, Shah S, Ganguly S, Zu Y, He C, Li Z. Classification of Acute Myeloid Leukemia by Cell-Free DNA 5-Hydroxymethylcytosine. Genes. 2023; 14(6):1180. https://doi.org/10.3390/genes14061180
Chicago/Turabian StyleShao, Jianming, Shilpan Shah, Siddhartha Ganguly, Youli Zu, Chuan He, and Zejuan Li. 2023. "Classification of Acute Myeloid Leukemia by Cell-Free DNA 5-Hydroxymethylcytosine" Genes 14, no. 6: 1180. https://doi.org/10.3390/genes14061180
APA StyleShao, J., Shah, S., Ganguly, S., Zu, Y., He, C., & Li, Z. (2023). Classification of Acute Myeloid Leukemia by Cell-Free DNA 5-Hydroxymethylcytosine. Genes, 14(6), 1180. https://doi.org/10.3390/genes14061180