


Genetic Origin and Introgression Pattern of Pingliang Red Cattle Revealed Using Genome-Wide SNP Analyses

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples Collection and Genotyping
2.2. Quality Control

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed | Abbr. | Chip Type | Geographical Region | Number of Pre-QC(N) | Number of Post-QC(N) | Source |
|---|---|---|---|---|---|---|
| Pingliang red | PLC | 100 K | Gansu, China | 122 | 122 | this study |
| Zaosheng | ZAS | 770 K | Gansu, China | 10 | 10 | this study |
| Qinchuan | QIC | 50 K | Shaanxi, China | 15 | 15 | Gao et al. [13] |
| 770 K | 16 | 16 | this study | |||
| Jinnan | JIN | 50 K | Shanxi, China | 14 | 13 | Gao et al. [13] |
| 770 K | 14 | 14 | Zhang et al. [22] | |||
| Mongolian | MGX | 50 K | Inner Mongolia Autonomous Region, China | 31 | 30 | Gao et al. [13] |
| Yanbian | YAB | 50 K | Jilin, China | 26 | 26 | Gao et al. [13] |
| Luxi | LUX | 50 K | Shandong, China | 11 | 11 | Gao et al. [13] |
| Nanyang | NAY | 50 K | Henan, China | 23 | 23 | Gao et al. [13] |
| Jian | JIA | 50 K | Jiangxi, China | 18 | 16 | Zhang et al. [22] |
| Leiqiong | LEQ | 50 K | Guangdong, China | 12 | 12 | Zhang et al. [22] |
| Limousin | LMS | 50 K | France | 21 | 21 | this study |
| 100 K | 9 | 9 | Gautier et al. [10] | |||
| South Devon | SDE | 100 K | England | 8 | 7 | this study |
| Red Angus | RAG | 50 K | England | 19 | 19 | this study |
| 100 K | 7 | 7 | Gao et al. [13] | |||
| Hanwoo | HAN | 50 K | Korea | 8 | 8 | Decker et al. [15] |
| Gir | GIR | 50 K | India | 24 | 24 | http://www.illumina.com/applications/agriculture.ilmn (accessed on 12 May 2022) |
| Nellore | NEL | 50 K | Brazilian | 21 | 21 | Matukumalli et al. [16] |
| Banteng | BLI | 50 K | Indonesia | 20 | 19 | Gao et al. [13] |
| Total | 449 | 443 |
2.3. Principal Component and Population Structure Analysis
2.4. Phylogenetic Tree Analysis
2.5. Identity by Descent Analysis
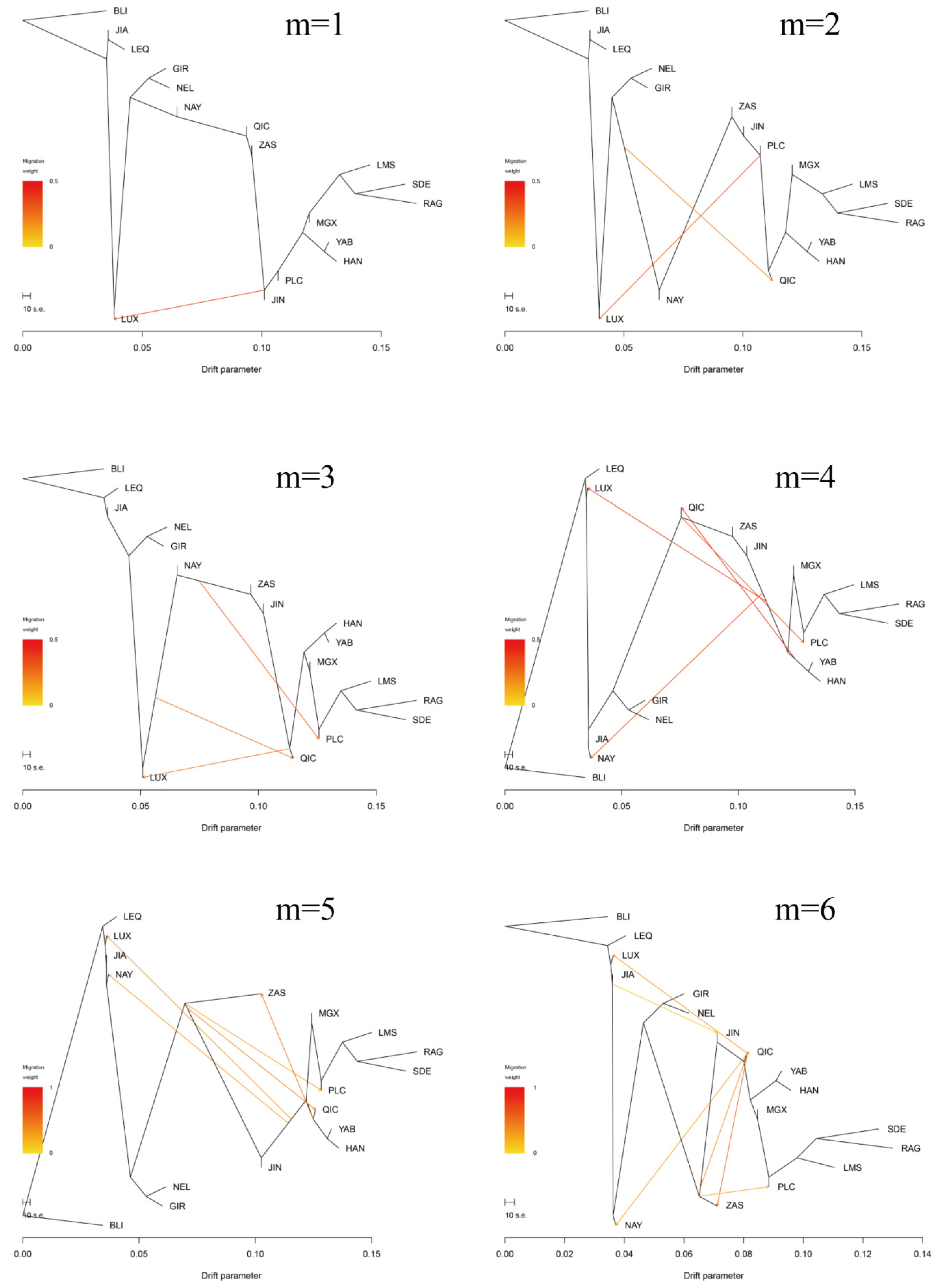
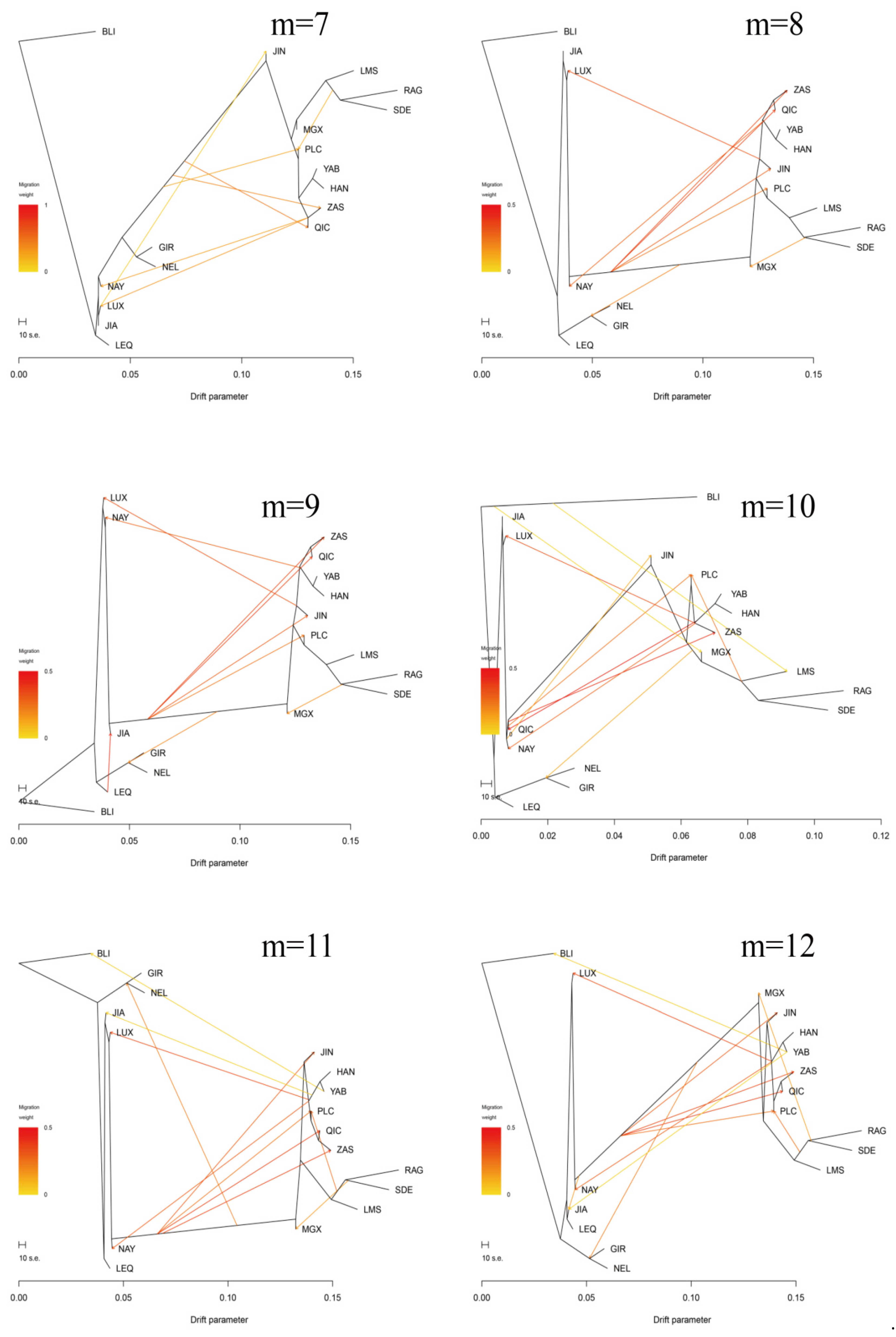
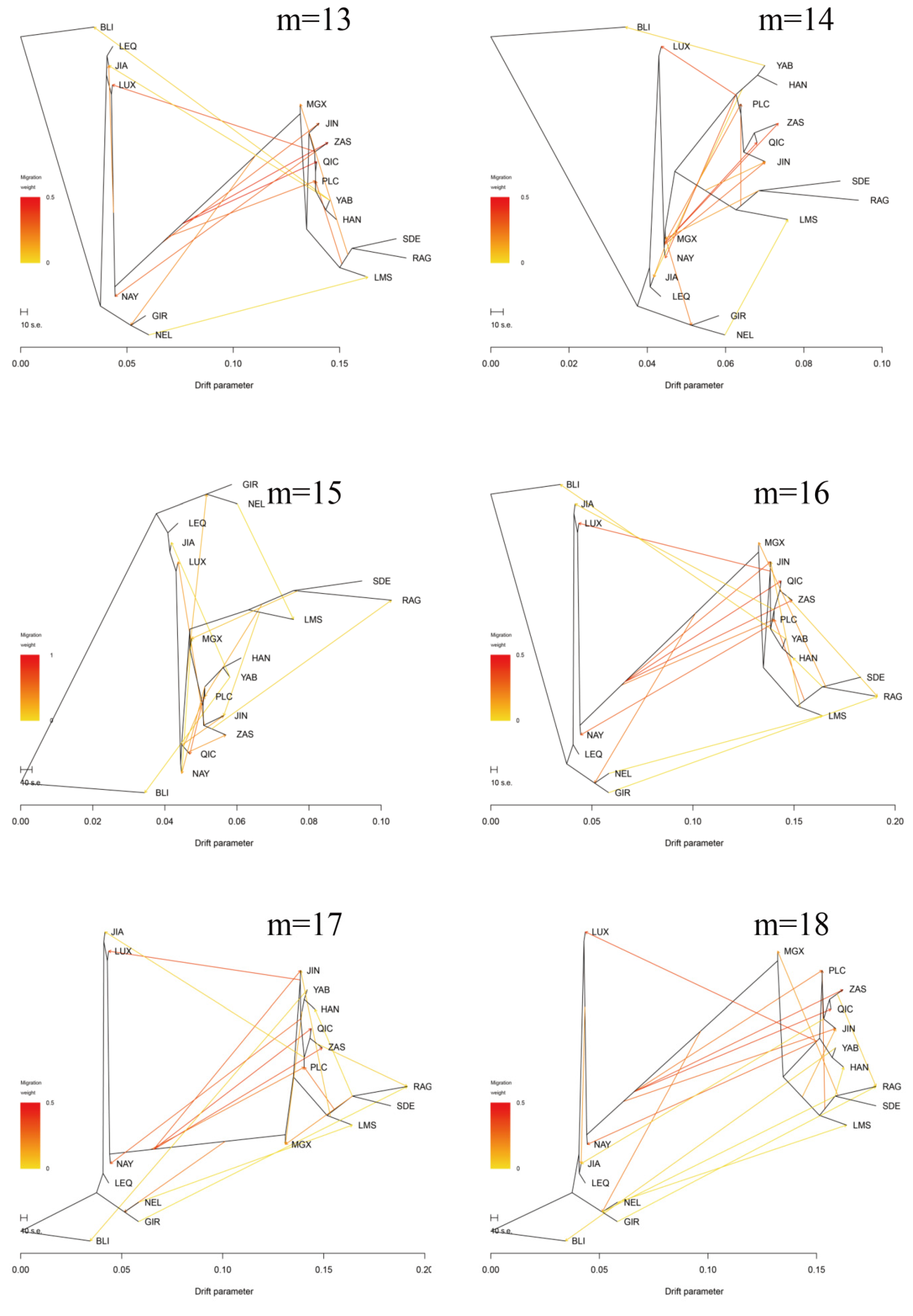
2.6. TreeMix Analysis
3. Results
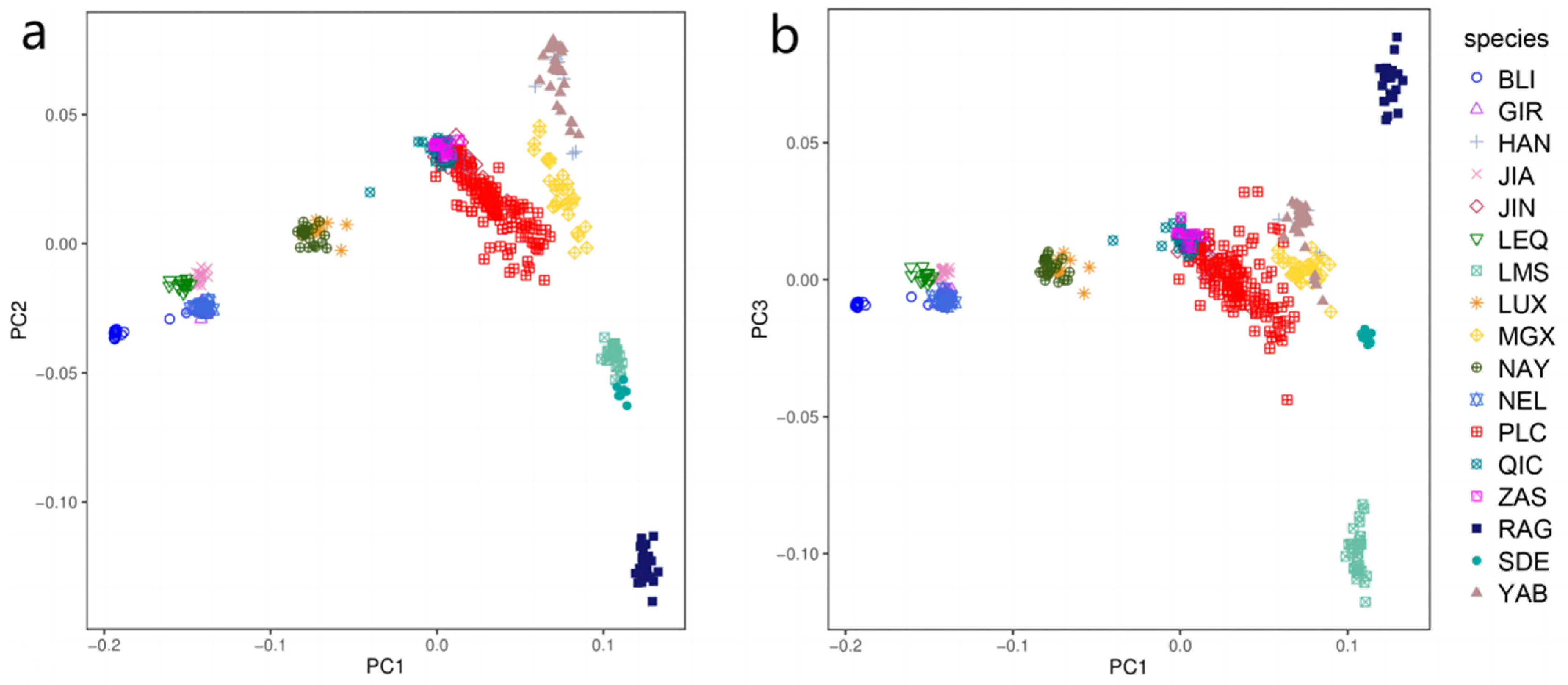
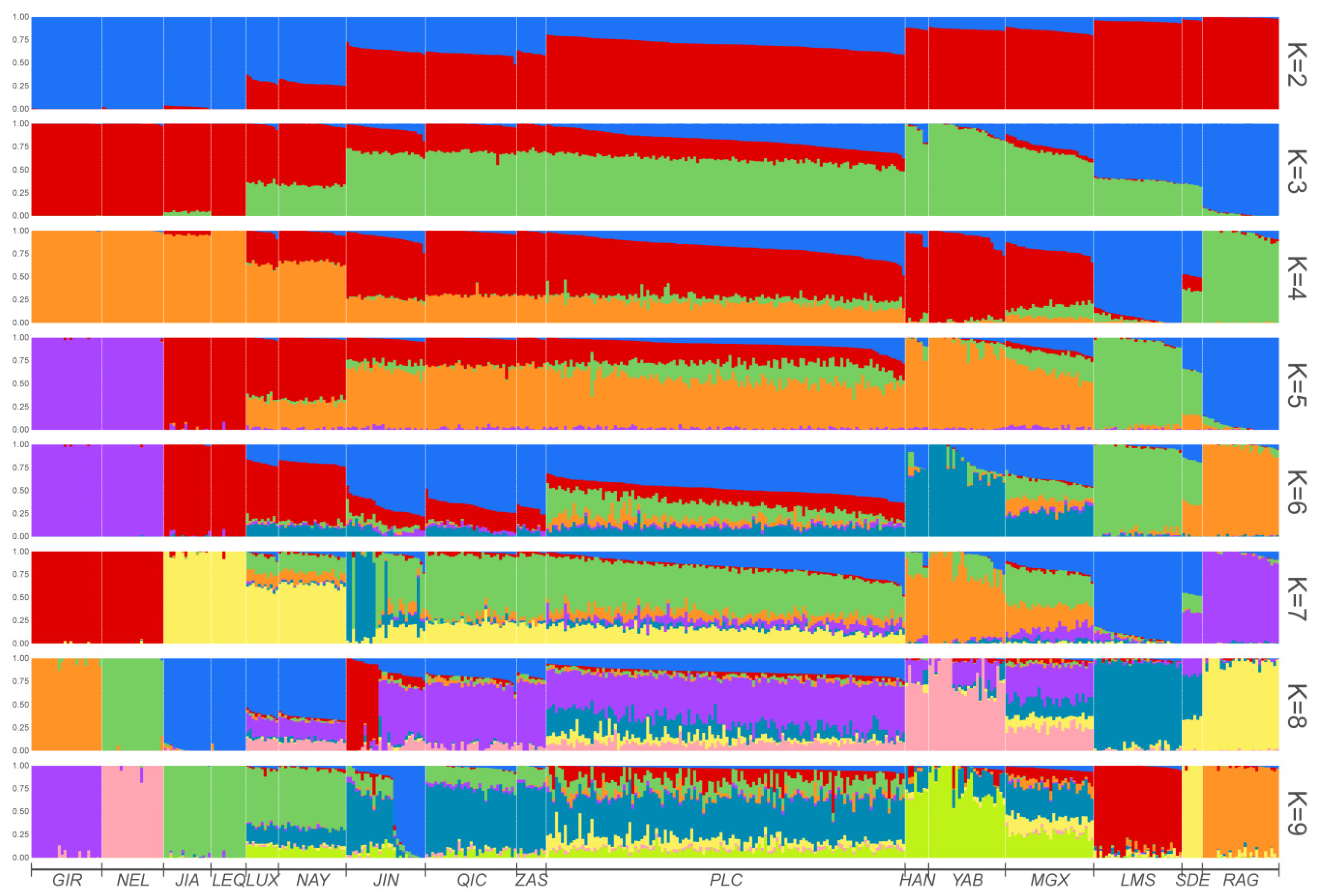
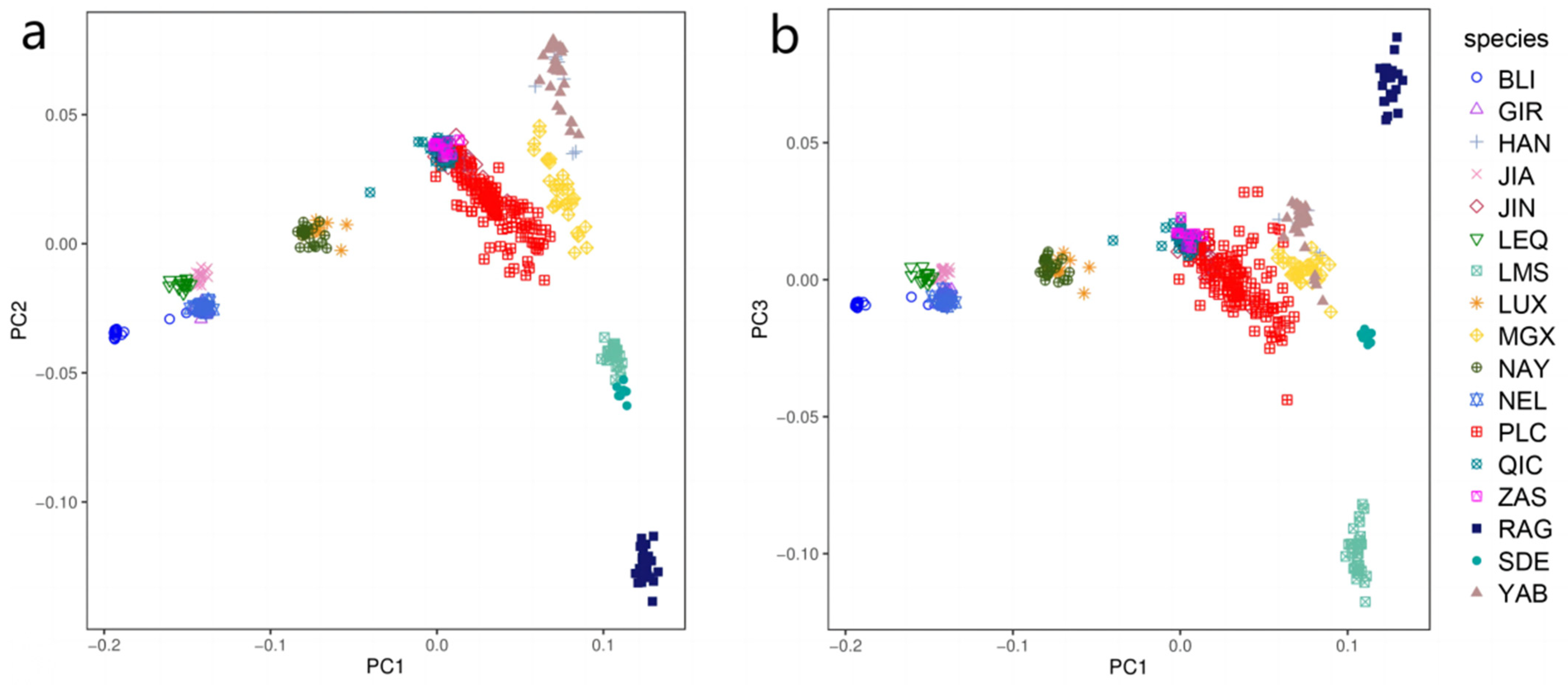
3.1. Principal Component and Population Structure Analysis
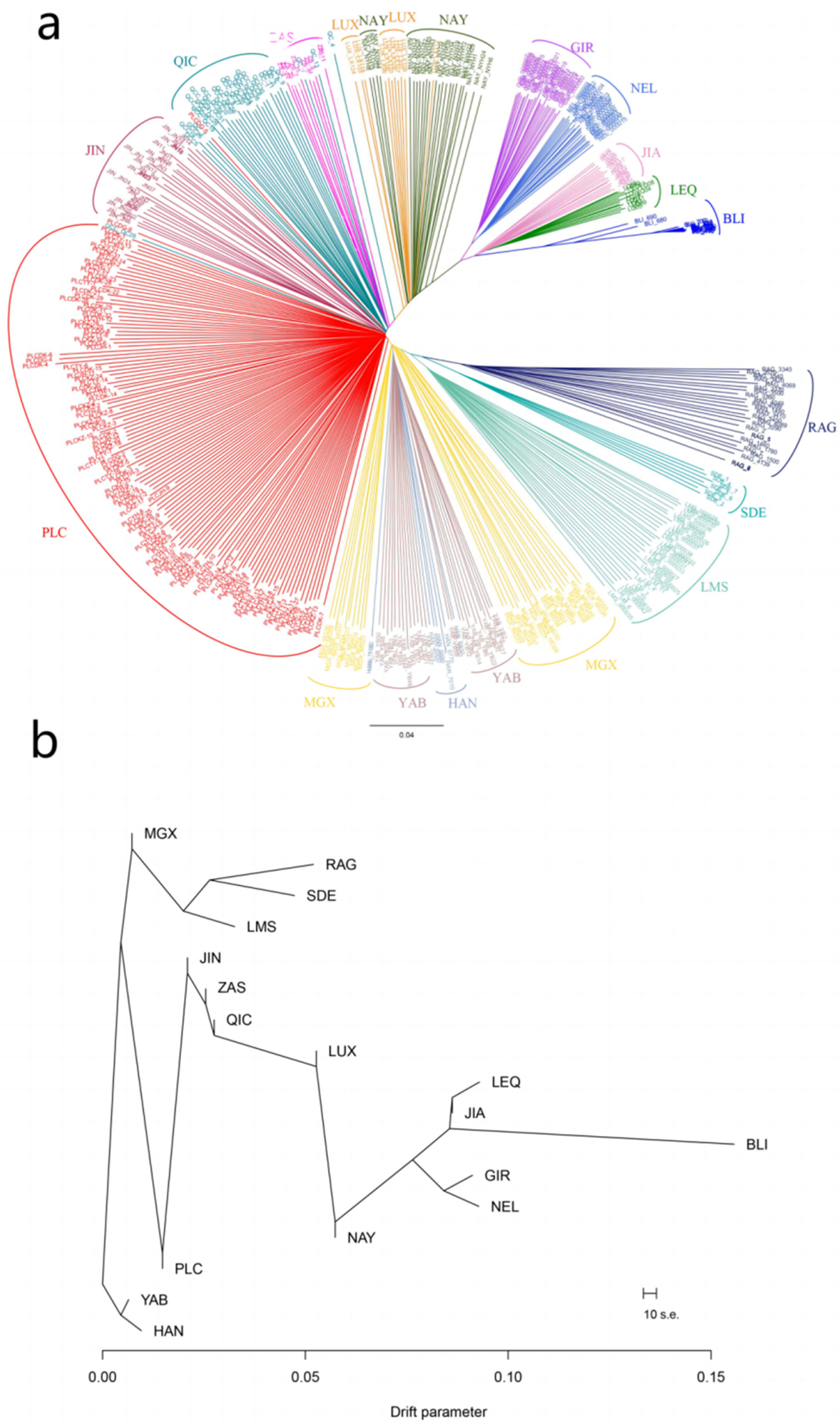
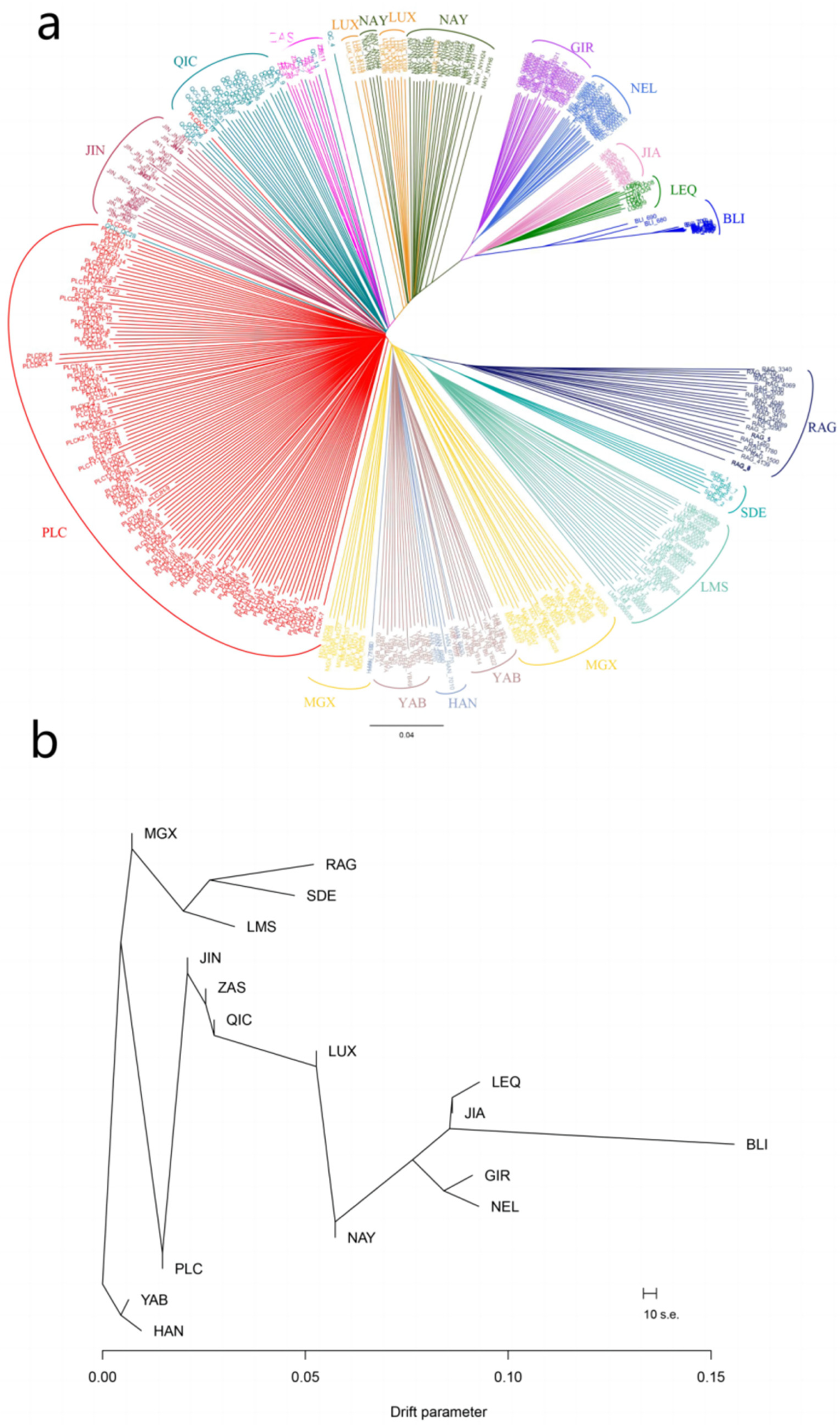
3.2. Phylogenetic Analysis of Pingliang Red Cattle with Other Breeds
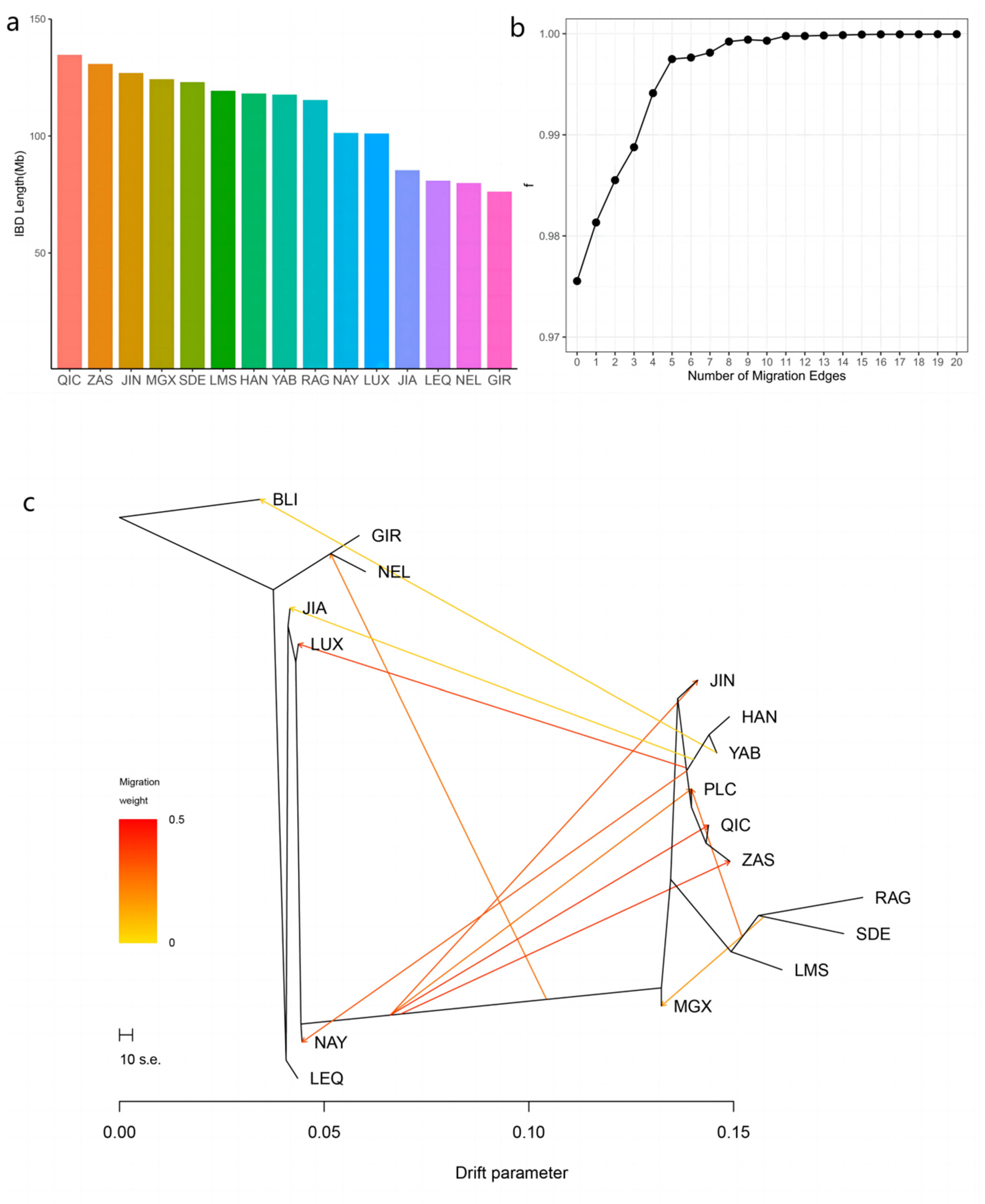
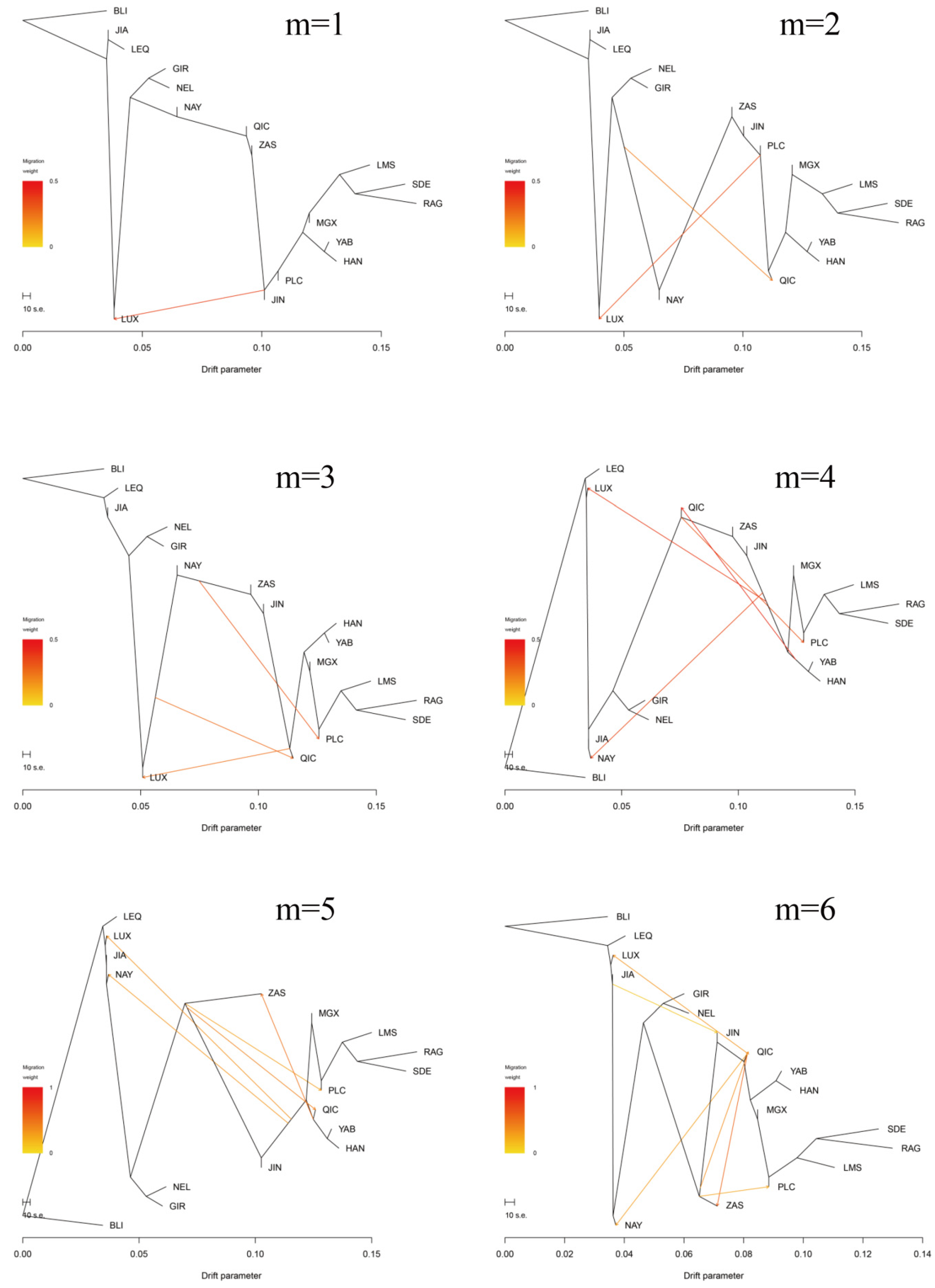
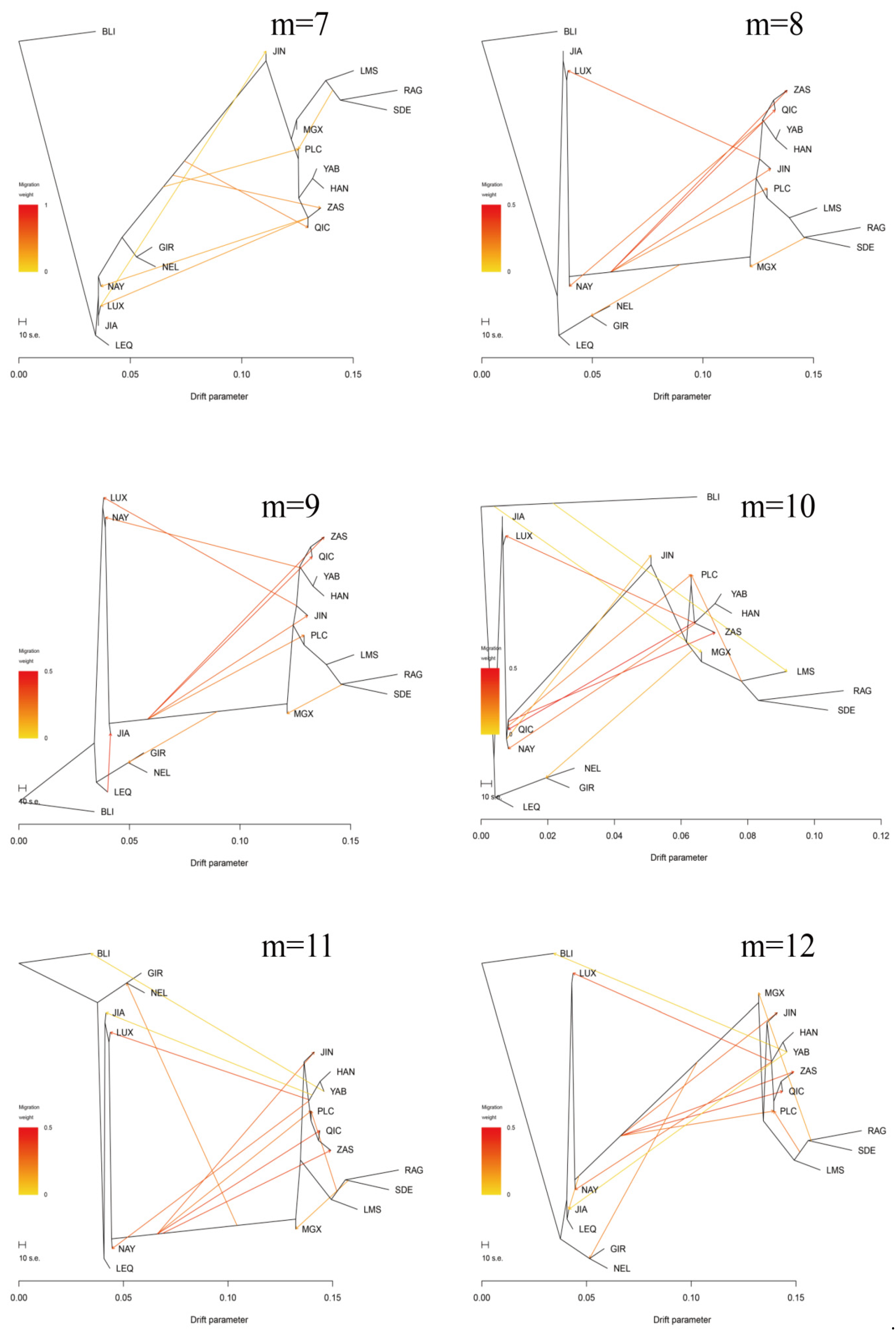
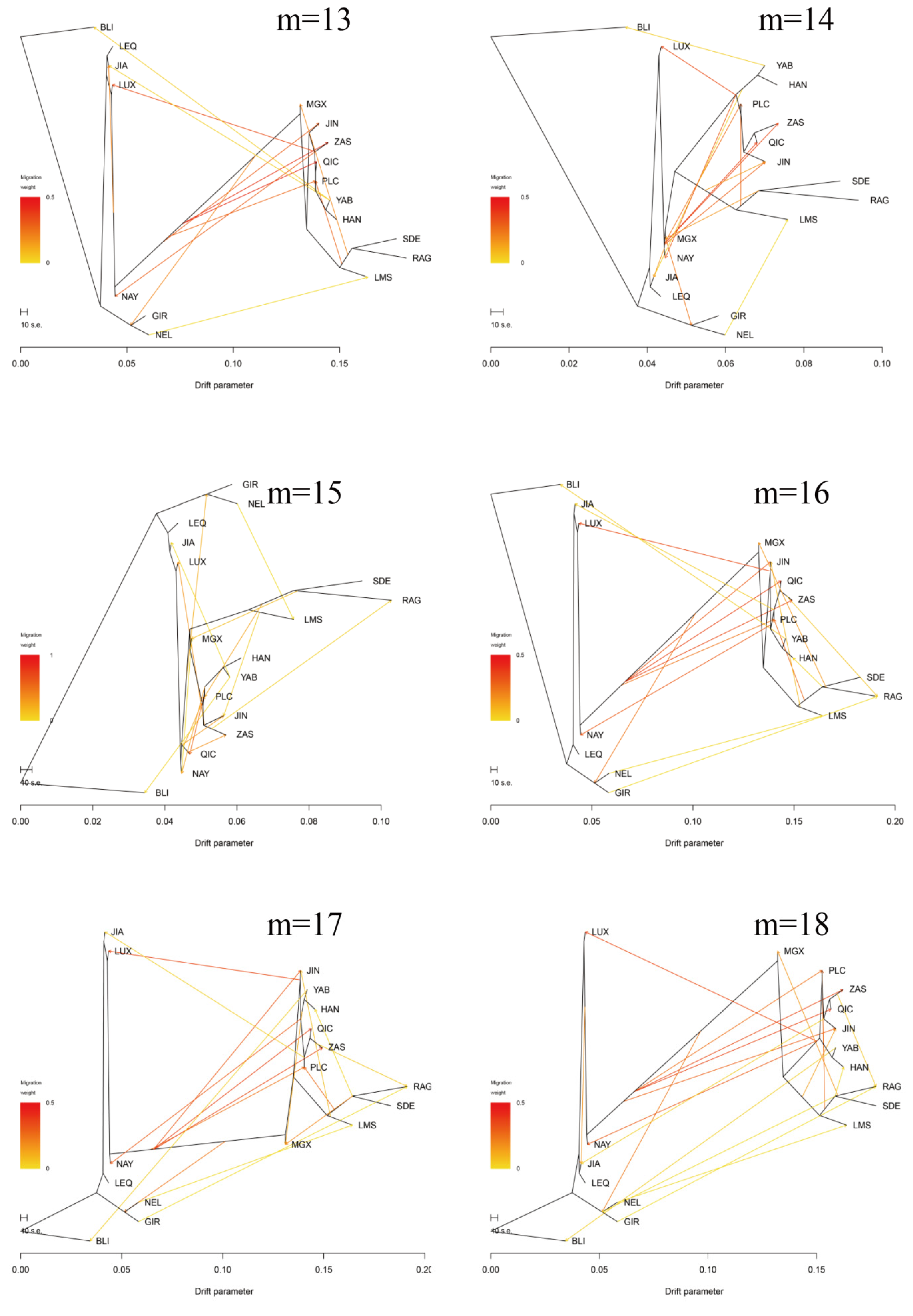
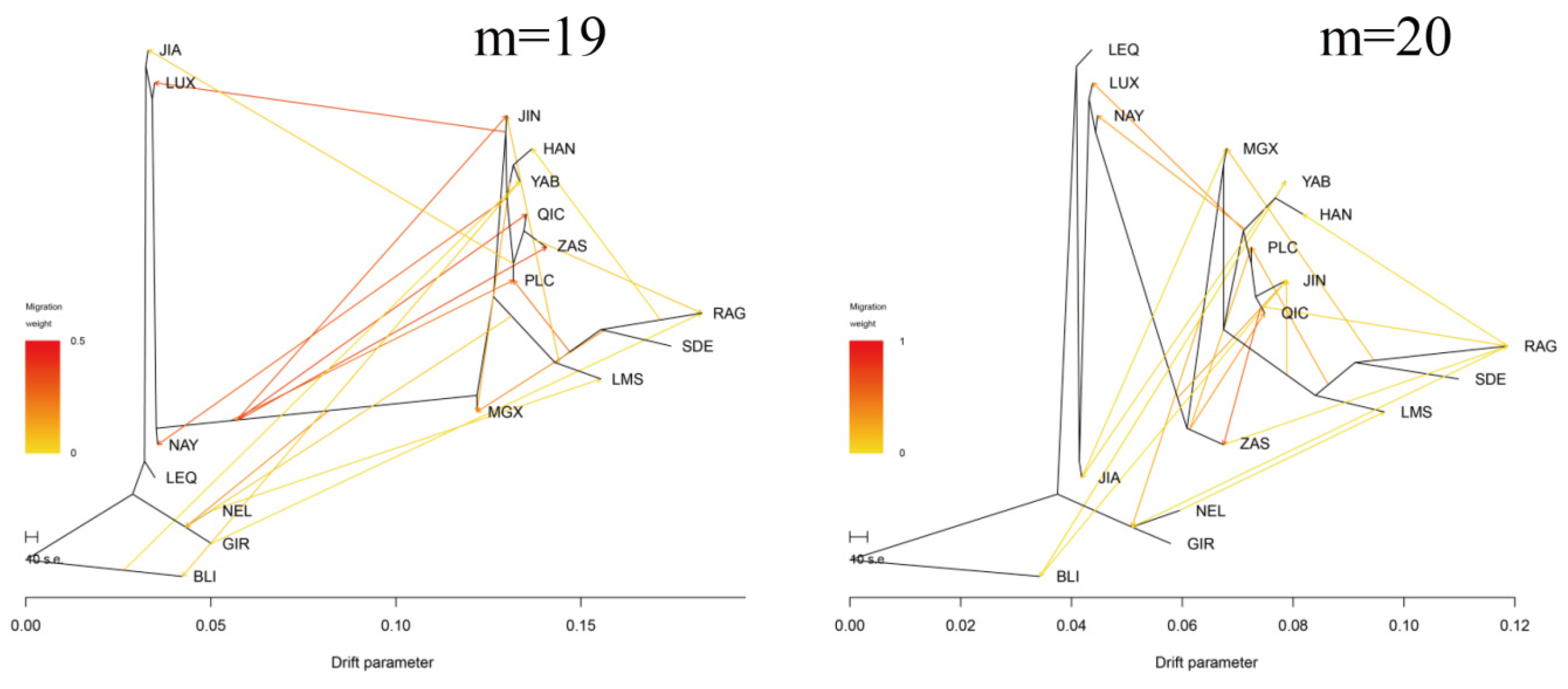
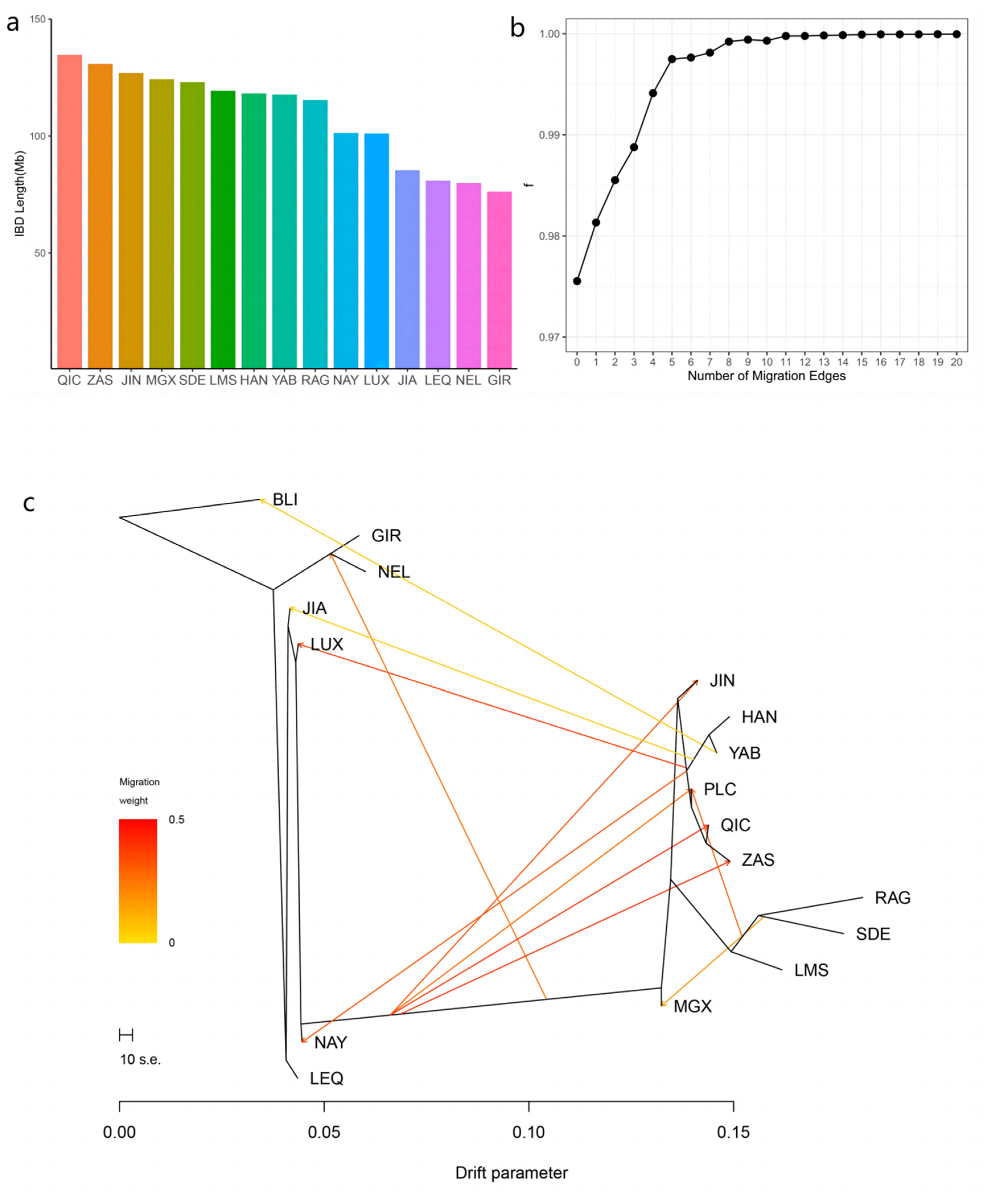
3.3. Introgression Pattern of the Pingliang Red Cattle
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Li, R.S. Research on “Pingliang Red cattle” Industrial Development. Chin. Cattle Sci. 2018, 44, 64–69. [Google Scholar]
- Zhao, S.G.; Li, W.B.; Chen, F.G.; Li, S.L.; Zhang, C.Q.; Wu, J.P. Maternal Genetic Background of Pingliang Native Cattle. Chin. J. Zool. 2013, 48, 109–117. [Google Scholar]
- Liu, L.; Zhao, S.G.; Cai, Y.; Lei, Z.M. The Genetic Diversity of Zaosheng Native Cattle (Bos taurus) and Associated Crossbred Population. J. Agric. Biotechnol. 2014, 22, 317–325. [Google Scholar]
- Cai, Y.; Jiao, T.; Lei, Z.; Liu, L.; Zhao, S. Maternal genetic and phylogenetic characteristics of domesticated cattle in northwestern China. PLoS ONE 2018, 13, e0209645. [Google Scholar] [CrossRef] [PubMed]
- Akey, J.M.; Ruhe, A.L.; Akey, D.T.; Wong, A.K.; Connelly, C.F.; Madeoy, J.; Nicholas, T.J.; Neff, M.W. Tracking footprints of artificial selection in the dog genome. Proc. Natl. Acad. Sci. USA 2010, 107, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Begun, D.J.; Holloway, A.K.; Stevens, K.; Hillier, L.W.; Poh, Y.P.; Hahn, M.W.; Nista, P.M.; Jones, C.D.; Kern, A.D.; Dewey, C.N.; et al. Population genomics: Whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 2007, 5, e310. [Google Scholar] [CrossRef] [PubMed]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef]
- Neto, L.R.P.; Barendse, W. Effect of SNP origin on analyses of genetic diversity in cattle. Anim. Prod. Sci. 2010, 50, 792–800. [Google Scholar] [CrossRef]
- Bovine HapMap Consortium; Gibbs, R.A.; Taylor, J.F.; Van Tassell, C.P.; Barendse, W.; Eversole, K.A.; Gill, C.A.; Green, R.D.; Hamernik, D.L.; Kappes, S.M. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 2009, 324, 528–532. [Google Scholar]
- Gautier, M.; Laloe, D.; Moazami-Goudarzi, K. Insights into the genetic history of French cattle from dense SNP data on 47 worldwide breeds. PLoS ONE 2010, 5, e13038. [Google Scholar] [CrossRef]
- Dadi, H.; Tibbo, M.; Takahashi, Y.; Nomura, K.; Hanada, H.; Amano, T. Microsatellite analysis reveals high genetic diversity but low genetic structure in Ethiopian indigenous cattle populations. Anim. Genet. 2008, 39, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, M.D.; King, J.M.; Loupis, A.K.; Spencer, P.B.; Taylor, A.C.; Pope, L.C.; Hall, G.P. Unprecedented low levels of genetic variation and inbreeding depression in an island population of the black-footed rock-wallaby. Conserv. Biol. 1999, 13, 531–541. [Google Scholar] [CrossRef]
- Gao, Y.; Gautier, M.; Ding, X.; Zhang, H.; Wang, Y.; Wang, X.; Faruque, M.O.; Li, J.; Ye, S.; Gou, X.; et al. Species composition and environmental adaptation of indigenous Chinese cattle. Sci. Rep. 2017, 7, 16196. [Google Scholar] [CrossRef]
- Xu, L.; Yang, L.; Zhu, B.; Zhang, W.; Wang, Z.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Liu, G.E.; et al. Genome-wide scan reveals genetic divergence and diverse adaptive selection in Chinese local cattle. BMC Genom. 2019, 20, 494. [Google Scholar] [CrossRef]
- Decker, J.E.; McKay, S.D.; Rolf, M.M.; Kim, J.; Molina Alcala, A.; Sonstegard, T.S.; Hanotte, O.; Gotherstrom, A.; Seabury, C.M.; Praharani, L.; et al. Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genet. 2014, 10, e1004254. [Google Scholar] [CrossRef] [PubMed]
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P.; O’Connell, J.; Moore, S.S.; Smith, T.P.; Sonstegard, T.S.; et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE 2009, 4, e5350. [Google Scholar] [CrossRef]
- Lai, S.J.; Liu, Y.P.; Liu, Y.X.; Li, X.W.; Yao, Y.G. Genetic diversity and origin of Chinese cattle revealed by mtDNA D-loop sequence variation. Mol. Phylogenet. Evol. 2006, 38, 146–154. [Google Scholar] [CrossRef]
- Long, J.C. The genetic structure of admixed populations. Genetics 1991, 127, 417–428. [Google Scholar] [CrossRef]
- Lv, F.H.; Agha, S.; Kantanen, J.; Colli, L.; Stucki, S.; Kijas, J.W.; Joost, S.; Li, M.H.; Ajmone Marsan, P. Adaptations to climate-mediated selective pressures in sheep. Mol. Biol. Evol. 2014, 31, 3324–3343. [Google Scholar] [CrossRef]
- Ma, H.; Wang, S.; Zeng, G.; Guo, J.; Guo, M.; Dong, X.; Hua, G.; Liu, Y.; Wang, M.; Ling, Y.; et al. The Origin of a Coastal Indigenous Horse Breed in China Revealed by Genome-Wide SNP Data. Genes 2019, 10, 241. [Google Scholar] [CrossRef]
- Han, L.; Abney, M. Identity by descent estimation with dense genome-wide genotype data. Genet. Epidemiol. 2011, 35, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, Y.; Wang, X.; Jiang, Q.; Zhao, H.; Wang, J.; Ju, Z.; Yang, L.; Gao, Y.; Wei, X.; et al. Population Structure, and Selection Signatures Underlying High-Altitude Adaptation Inferred from Genome-Wide Copy Number Variations in Chinese Indigenous Cattle. Front. Genet. 2019, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Kijas, J.W.; Heaton, M.P.; McEwan, J.C.; Coltman, D.W. Consistent divergence times and allele sharing measured from cross-species application of SNP chips developed for three domestic species. Mol. Ecol. Resour. 2012, 12, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Msalya, G.; Kim, E.S.; Laisser, E.L.; Kipanyula, M.J.; Karimuribo, E.D.; Kusiluka, L.J.; Chenyambuga, S.W.; Rothschild, M.F. Determination of Genetic Structure and Signatures of Selection in Three Strains of Tanzania Shorthorn Zebu, Boran and Friesian Cattle by Genome-Wide SNP Analyses. PLoS ONE 2017, 12, e0171088. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Pritchard, J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef]
- Rosenberg, N.A.; Nordborg, M. A general population-genetic model for the production by population structure of spurious genotype-phenotype associations in discrete, admixed or spatially distributed populations. Genetics 2006, 173, 1665–1678. [Google Scholar] [CrossRef]
- Qanbari, S.; Gianola, D.; Hayes, B.; Schenkel, F.; Miller, S.; Moore, S.; Thaller, G.; Simianer, H. Application of site and haplotype-frequency based approaches for detecting selection signatures in cattle. BMC Genom. 2011, 12, 318. [Google Scholar] [CrossRef]
- Urbinati, I.; Stafuzza, N.B.; Oliveira, M.T.; Chud, T.C.; Higa, R.H.; Regitano, L.C.; de Alencar, M.M.; Buzanskas, M.E.; Munari, D.P. Selection signatures in Canchim beef cattle. J. Anim. Sci. Biotechnol. 2016, 7, 29. [Google Scholar] [CrossRef]
- Chen, N.; Cai, Y.; Chen, Q.; Li, R.; Wang, K.; Huang, Y.; Hu, S.; Huang, S.; Zhang, H.; Zheng, Z.; et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 2018, 9, 2337. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, D.; Guo, X.; Yang, W.; Sun, J.; Wang, D.; Sourdille, P.; Zhang, A. Investigation of genetic diversity and population structure of common wheat cultivars in northern China using DArT markers. BMC Genet. 2011, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Calus, M.P.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G. Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. BMC Genet. 2015, 16, 88. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya, Y.T.; do Carmo, A.S.; Carvalheiro, R.; Neves, H.H.; Matos, M.C.; Zavarez, L.B.; Perez O’Brien, A.M.; Solkner, J.; McEwan, J.C.; Cole, J.B.; et al. Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, e72. [Google Scholar]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 1358–1370. [Google Scholar] [CrossRef]
- Zhao, P.; Yu, Y.; Feng, W.; Du, H.; Yu, J.; Kang, H.; Zheng, X.; Wang, Z.; Liu, G.E.; Ernst, C.W.; et al. Evidence of evolutionary history and selective sweeps in the genome of Meishan pig reveals its genetic and phenotypic characterization. Gigascience 2018, 7, giy058. [Google Scholar] [CrossRef]
- Traspov, A.; Deng, W.; Kostyunina, O.; Ji, J.; Shatokhin, K.; Lugovoy, S.; Zinovieva, N.; Yang, B.; Huang, L. Population structure and genome characterization of local pig breeds in Russia, Belorussia, Kazakhstan and Ukraine. Genet. Sel. Evol. 2016, 48, 16. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Ma, J.; Wang, J.; Zhang, L.; Hu, J.; Ma, M.; Xu, L.; Chen, Y.; Zhu, B.; Wang, Z.; et al. Genetic Origin and Introgression Pattern of Pingliang Red Cattle Revealed Using Genome-Wide SNP Analyses. Genes 2023, 14, 2198. https://doi.org/10.3390/genes14122198
Wang Y, Ma J, Wang J, Zhang L, Hu J, Ma M, Xu L, Chen Y, Zhu B, Wang Z, et al. Genetic Origin and Introgression Pattern of Pingliang Red Cattle Revealed Using Genome-Wide SNP Analyses. Genes. 2023; 14(12):2198. https://doi.org/10.3390/genes14122198
Chicago/Turabian StyleWang, Yuanqing, Jun Ma, Jing Wang, Lupei Zhang, Junwei Hu, Minghao Ma, Lingyang Xu, Yan Chen, Bo Zhu, Zezhao Wang, and et al. 2023. "Genetic Origin and Introgression Pattern of Pingliang Red Cattle Revealed Using Genome-Wide SNP Analyses" Genes 14, no. 12: 2198. https://doi.org/10.3390/genes14122198
APA StyleWang, Y., Ma, J., Wang, J., Zhang, L., Hu, J., Ma, M., Xu, L., Chen, Y., Zhu, B., Wang, Z., Gao, H., Li, J., & Gao, X. (2023). Genetic Origin and Introgression Pattern of Pingliang Red Cattle Revealed Using Genome-Wide SNP Analyses. Genes, 14(12), 2198. https://doi.org/10.3390/genes14122198