
Characterization of the Complete Mitochondrial Genome and Phylogenetic Analyses of Eurytrema coelomaticum (Trematoda: Dicrocoeliidae)

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of Eurytrema coelomaticum
2.2. DNA Extraction and Sequencing
2.3. Genome Sequence Assembly and Analysis
2.4. Genome Annotation
2.5. Phylogenetic Analysis
3. Results
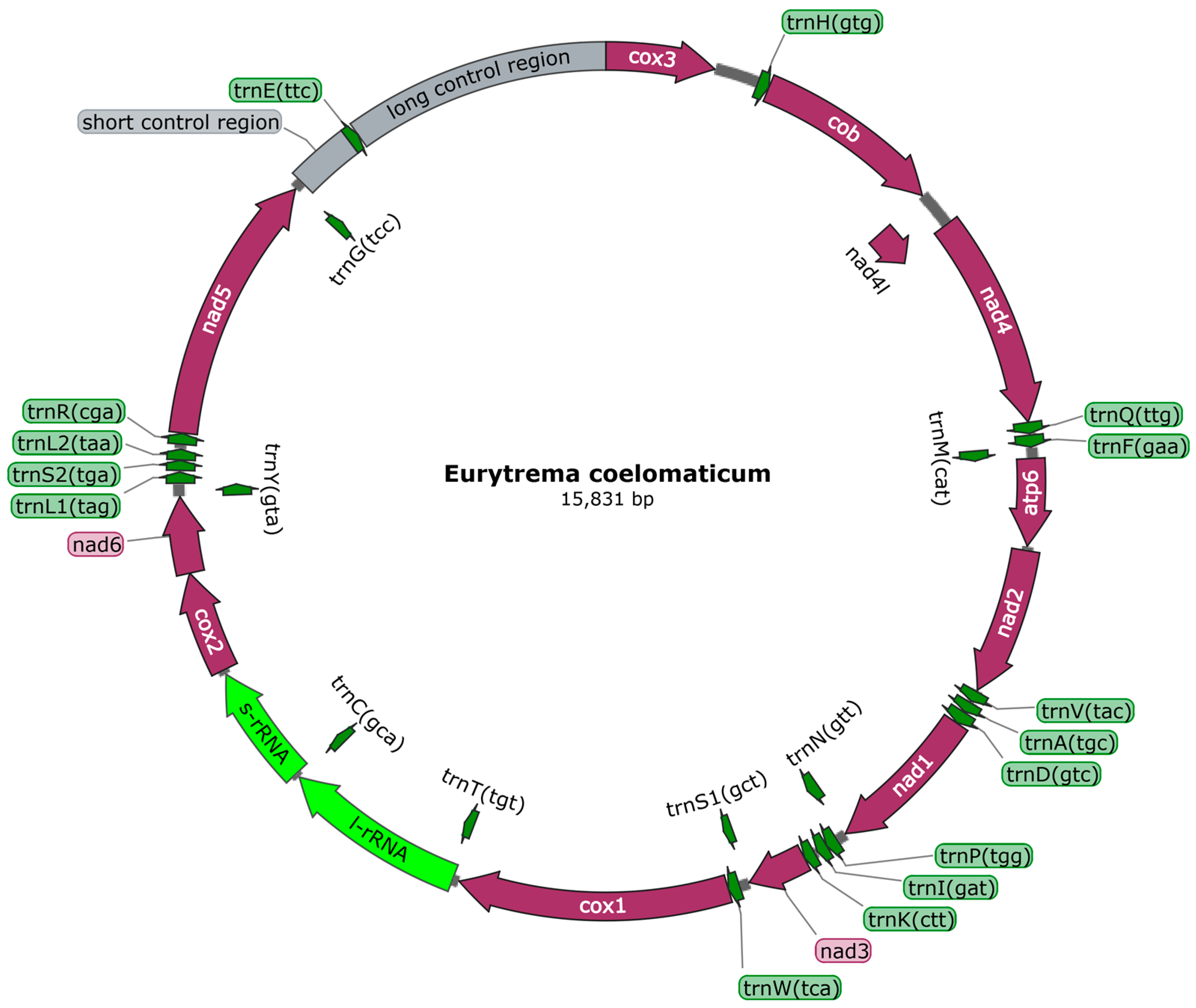
3.1. Mitogenome Structure and Nucleotide Composition
3.2. Protein-Coding Genes and Codon Usage
3.3. tRNA and rRNA Genes
3.4. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ishii, Y.; Koga, M.; Fujino, T.; Higo, H.; Ishibashi, J.; Oka, K.; Saito, S. Human infection with the pancreas fluke Eurytrema pancreaticum. Am. J. Trop. Med. Hyg. 1983, 32, 1019–1022. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, D.E.R.; Barbosa, E.F.G.; Wilson, T.M.; Machado, M.; Oliveira, W.J.; Duarte, M.A.; Scalon, M.C.; Câmara, A.C.L.; Lux Hoppe, E.G.; Paludo, G.R.; et al. Eurytrema coelomaticum natural infection in small ruminants: A neglected condition. Parasitology 2021, 148, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Ilha, M.R.; Loretti, A.P.; Reis, A.C. Wasting and mortality in beef cattle parasitized by Eurytrema coelomaticum in the State of Paraná, southern Brazil. Vet. Parasitol. 2005, 133, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Rojo-Vázquez, F.A.; Meana, A.; Valcárcel, F.; Martínez-Valladares, M. Update on trematode infections in sheep. Vet. Parasitol. 2012, 189, 15–38. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, U.K.; Ichikawa-Seki, M.; Hayashi, K.; Itagaki, T. Morphological and molecular characterization of Eurytrema cladorchis parasitizing cattle (Bos indicus) in Bangladesh. Parasitol. Res. 2015, 114, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Jones, A. Eurytrema cladorchis Chin, Li & Wei, 1965 (Trematoda: Dicrocoeliidae), a little known species from China and Nepal. Syst. Parasitol. 1985, 7, 43–45. [Google Scholar] [CrossRef]
- Song, M.; Zhang, L. Veterinary Parasitology; China Science Publishing Media Ltd.: Beijing, China, 2009. (In Chinese) [Google Scholar]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef]
- Wang, P.; Yang, H.F.; Zhou, W.C.; Hwang, C.C.; Zhang, W.H.; Qian, Z.X. The mitochondrial genome of the land snail Camaena cicatricosa (Müller, 1774) (Stylommatophora, Camaenidae): The first complete sequence in the family Camaenidae. ZooKeys 2014, 451, 33–48. [Google Scholar] [CrossRef]
- Leite, K.G.; Lopes-Torres, E.J.; Souza, J.G.R.; Neves, R.H.; Gomes, D.C.; Machado-Silva, J.R. Eurytrema coelomaticum: Updated morphology of adult worms using advanced microscopy experiments. J. Helminthol. 2020, 94, e122. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, L.; Tian, S.; Zhang, B.; Liang, C.; Huang, F.; Liu, Q.; Zhang, H. Morphological Observation and Molecular Identification of Eurytrema coelmaticum in Goats. Progress. Vet. Med. 2019, 40, 34–38. (In Chinese) [Google Scholar] [CrossRef]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 32, 1933–1942. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Chernomor, O.; von Haeseler, A.; Minh, B.Q. Terrace Aware Data Structure for Phylogenomic Inference from Supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Minh, B.Q.; Susko, E.; Roger, A.J. Modeling Site Heterogeneity with Posterior Mean Site Frequency Profiles Accelerates Accurate Phylogenomic Estimation. Syst. Biol. 2018, 67, 216–235. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Littlewood, D.T.; Lockyer, A.E.; Webster, B.L.; Johnston, D.A.; Le, T.H. The complete mitochondrial genomes of Schistosoma haematobium and Schistosoma spindale and the evolutionary history of mitochondrial genome changes among parasitic flatworms. Mol. Phylogenet. Evol. 2006, 39, 452–467. [Google Scholar] [CrossRef]
- Liu, G.H.; Gasser, R.B.; Young, N.D.; Song, H.Q.; Ai, L.; Zhu, X.Q. Complete mitochondrial genomes of the ‘intermediate form’ of Fasciola and Fasciola gigantica, and their comparison with F. hepatica. Parasites Vectors 2014, 7, 150. [Google Scholar] [CrossRef]
- Jones, B.P.; Norman, B.F.; Borrett, H.E.; Attwood, S.W.; Mondal, M.M.H.; Walker, A.J.; Webster, J.P.; Rajapakse, R.; Lawton, S.P. Divergence across mitochondrial genomes of sympatric members of the Schistosoma indicum group and clues into the evolution of Schistosoma spindale. Sci. Rep. 2020, 10, 2480. [Google Scholar] [CrossRef]
- Le, T.H.; Humair, P.F.; Blair, D.; Agatsuma, T.; Littlewood, D.T.; McManus, D.P. Mitochondrial gene content, arrangement and composition compared in African and Asian schistosomes. Mol. Biochem. Parasitol. 2001, 117, 61–71. [Google Scholar] [CrossRef]
- Le, T.H.; Blair, D.; McManus, D.P. Mitochondrial genomes of parasitic flatworms. Trends Parasitol. 2002, 18, 206–213. [Google Scholar] [CrossRef]
- Chang, Q.C.; Liu, G.H.; Gao, J.F.; Zheng, X.; Zhang, Y.; Duan, H.; Yue, D.M.; Fu, X.; Su, X.; Gao, Y.; et al. Sequencing and characterization of the complete mitochondrial genome from the pancreatic fluke Eurytrema pancreaticum (Trematoda: Dicrocoeliidae). Gene 2016, 576, 160–165. [Google Scholar] [CrossRef]
- Kinkar, L.; Young, N.D.; Sohn, W.M.; Stroehlein, A.J.; Korhonen, P.K.; Gasser, R.B. First record of a tandem-repeat region within the mitochondrial genome of Clonorchis sinensis using a long-read sequencing approach. PLoS Negl. Trop. Dis. 2020, 14, e0008552. [Google Scholar] [CrossRef] [PubMed]
- Kinkar, L.; Gasser, R.B.; Webster, B.L.; Rollinson, D.; Littlewood, D.T.J.; Chang, B.C.H.; Stroehlein, A.J.; Korhonen, P.K.; Young, N.D. Nanopore Sequencing Resolves Elusive Long Tandem-Repeat Regions in Mitochondrial Genomes. Int. J. Mol. Sci. 2021, 22, 1811. [Google Scholar] [CrossRef]
- Tkach, V.; Pawlowski, J.; Mariaux, J. Phylogenetic analysis of the suborder plagiorchiata (Platyhelminthes, Digenea) based on partial lsrDNA sequences. Int. J. Parasitol. 2000, 30, 83–93. [Google Scholar] [CrossRef]
- Figueira, G.F.; Oliveira, V.H.; Taroda, A.; Alfieri, A.A.; Headley, S.A. Molecular characterization of Eurytrema coelomaticum in cattle from Paraná, Brazil. Rev. Bras. De Parasitol. Vet. Braz. J. Vet. Parasitol. 2014, 23, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.B.; Wang, X.Y.; Lou, Z.Z.; Li, L.; Blair, D.; Yin, H.; Cai, J.Z.; Dai, X.L.; Lei, M.T.; Zhu, X.Q.; et al. The mitochondrial genome of Paramphistomum cervi (Digenea), the first representative for the family Paramphistomidae. PLoS ONE 2013, 8, e71300. [Google Scholar] [CrossRef]
- Liu, G.H.; Yan, H.B.; Otranto, D.; Wang, X.Y.; Zhao, G.H.; Jia, W.Z.; Zhu, X.Q. Dicrocoelium chinensis and Dicrocoelium dendriticum (Trematoda: Digenea) are distinct lancet fluke species based on mitochondrial and nuclear ribosomal DNA sequences. Mol. Phylogenet. Evol. 2014, 79, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.R.; Li, Y.; Gao, Y.; Qiu, Y.Y.; Jin, Z.H.; Gao, Z.Y.; Zhang, X.G.; An, Q.; Chang, Q.C.; Gao, J.F.; et al. The complete mitochondrial genome of Prosthogonimus cuneatus and Prosthogonimus pellucidus (Trematoda: Prosthogonimidae), their features and phylogenetic relationships in the superfamily Microphalloidea. Acta Trop. 2022, 232, 106469. [Google Scholar] [CrossRef]
- Briscoe, A.G.; Bray, R.A.; Brabec, J.; Littlewood, D.T. The mitochondrial genome and ribosomal operon of Brachycladium goliath (Digenea: Brachycladiidae) recovered from a stranded minke whale. Parasitol. Int. 2016, 65, 271–275. [Google Scholar] [CrossRef]
- Fu, Y.T.; Jin, Y.C.; Liu, G.H. The Complete Mitochondrial Genome of the Caecal Fluke of Poultry, Postharmostomum commutatum, as the First Representative from the Superfamily Brachylaimoidea. Front. Genet. 2019, 10, 1037. [Google Scholar] [CrossRef]
- Olson, P.D.; Cribb, T.H.; Tkach, V.V.; Bray, R.A.; Littlewood, D.T.J. Phylogeny and classification of the Digenea (Platyhelminthes: Trematoda). Int. J. Parasitol. 2003, 33, 733–755. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regions | Strand | Size (bp) | T(U) (%) | C (%) | A (%) | G (%) | A+T (%) | G+C (%) | AT Skewness | GC Skewness |
|---|---|---|---|---|---|---|---|---|---|---|
| Full genome | + | 15,831 | 43.7 | 11.9 | 18.8 | 25.6 | 62.5 | 37.5 | −0.399 | 0.365 |
| PCGs | all | 10,302 | 47 | 11.7 | 15.8 | 25.5 | 62.8 | 37.2 | −0.497 | 0.37 |
| PCGs | + | 10,302 | 47 | 11.7 | 15.8 | 25.5 | 62.8 | 37.2 | −0.497 | 0.37 |
| tRNAs | all | 1476 | 36.8 | 13.9 | 21.1 | 28.2 | 57.9 | 42.1 | −0.27 | 0.34 |
| tRNAs | + | 1476 | 36.8 | 13.9 | 21.1 | 28.2 | 57.9 | 42.1 | −0.27 | 0.34 |
| rRNAs | all | 1820 | 38.2 | 12.4 | 22.6 | 26.8 | 60.8 | 39.2 | −0.256 | 0.366 |
| rRNAs | + | 1820 | 38.2 | 12.4 | 22.6 | 26.8 | 60.8 | 39.2 | −0.256 | 0.366 |
| 1st codon position | all | 3434 | 40.3 | 12.1 | 19.5 | 28 | 59.8 | 40.1 | −0.347 | 0.398 |
| 1st codon position | + | 3434 | 40.3 | 12.1 | 19.5 | 28 | 59.8 | 40.1 | −0.347 | 0.398 |
| 2nd codon position | all | 3434 | 46.8 | 16 | 16.3 | 21 | 63.1 | 37 | −0.484 | 0.135 |
| 2nd codon position | + | 3434 | 46.8 | 16 | 16.3 | 21 | 63.1 | 37 | −0.484 | 0.135 |
| 3rd codon position | all | 3434 | 53.9 | 7.1 | 11.6 | 27.5 | 65.5 | 34.6 | −0.647 | 0.59 |
| 3rd codon position | + | 3434 | 53.9 | 7.1 | 11.6 | 27.5 | 65.5 | 34.6 | −0.647 | 0.59 |
| atp6 | + | 519 | 52.8 | 11.6 | 11.2 | 24.5 | 64 | 36.1 | −0.651 | 0.358 |
| cox1 | + | 1620 | 44.4 | 13.8 | 16.6 | 25.2 | 61 | 39 | −0.456 | 0.293 |
| cox2 | + | 609 | 43 | 13.8 | 17.4 | 25.8 | 60.4 | 39.6 | −0.424 | 0.303 |
| cox3 | + | 651 | 46.7 | 12.3 | 16.4 | 24.6 | 63.1 | 36.9 | −0.479 | 0.333 |
| cytb | + | 1119 | 46.7 | 12.2 | 16.5 | 24.5 | 63.2 | 36.7 | −0.477 | 0.333 |
| nad1 | + | 933 | 45.3 | 10.5 | 16.6 | 27.5 | 61.9 | 38 | −0.464 | 0.448 |
| nad2 | + | 879 | 49.8 | 9.9 | 14.2 | 26.1 | 64 | 36 | −0.556 | 0.449 |
| nad3 | + | 357 | 50.4 | 8.4 | 15.1 | 26.1 | 65.5 | 34.5 | −0.538 | 0.512 |
| nad4 | + | 1278 | 48 | 10.9 | 13.7 | 27.4 | 61.7 | 38.3 | −0.556 | 0.431 |
| nad4L | + | 264 | 45.8 | 9.5 | 18.2 | 26.5 | 64 | 36 | −0.432 | 0.474 |
| nad5 | + | 1617 | 46.5 | 12.4 | 17.5 | 23.6 | 64 | 36 | −0.453 | 0.309 |
| nad6 | + | 456 | 50.7 | 9.4 | 13.6 | 26.3 | 64.3 | 35.7 | −0.577 | 0.472 |
| rrnL | + | 1098 | 39.9 | 11.9 | 22.3 | 25.9 | 62.2 | 37.8 | −0.283 | 0.369 |
| rrnS | + | 722 | 35.6 | 13.2 | 23.1 | 28.1 | 58.7 | 41.3 | −0.212 | 0.362 |
| SNCR | / | 371 | 41.5 | 11.6 | 27.2 | 19.7 | 69.7 | 31.3 | −0.208 | 0.259 |
| LNCR | / | 1564 | 35 | 11,6 | 31 | 22.4 | 66 | 34 | −0.061 | 0.318 |
| Feature | Position | Length (bp) | Initiation Codon | Stop Codon | Anticodon | Intergenic Nucleotide |
|---|---|---|---|---|---|---|
| cox3 | 1–651 | 651 | GTG | TAG | 266 | |
| trnH | 918–987 | 70 | GTG | 1 | ||
| cob | 989–2107 | 1119 | ATG | TAA | −1 | |
| nad4l | 2107–2370 | 264 | ATG | TAA | −40 | |
| nad4 | 2331–3608 | 1278 | ATG | TAG | 3 | |
| trnQ | 3612–3676 | 65 | TTG | 11 | ||
| trnF | 3688–3757 | 70 | GAA | −3 | ||
| trnM | 3755–3822 | 68 | CAT | 3 | ||
| atp6 | 3826–4344 | 519 | ATG | TAA | 27 | |
| nad2 | 4372–5250 | 879 | ATG | TAA | 10 | |
| trnV | 5261–5325 | 65 | TAC | 10 | ||
| trnA | 5336–5398 | 63 | TGC | 5 | ||
| trnD | 5404–5477 | 74 | GTC | 3 | ||
| nad1 | 5481–6413 | 933 | ATG | TAA | −29 | |
| trnN | 6385–6464 | 80 | GTT | 10 | ||
| trnP | 6475–6545 | 71 | TGG | 0 | ||
| trnI | 6546–6610 | 65 | GAT | 18 | ||
| trnK | 6629–6694 | 66 | CTT | 1 | ||
| nad3 | 6696–7052 | 357 | ATG | TAA | −12 | |
| trnS1 | 7041–7100 | 60 | GCT | 13 | ||
| trnW | 7114–7181 | 68 | TCA | 1 | ||
| cox1 | 7183–8802 | 1620 | ATG | TAG | 35 | |
| trnT | 8838–8904 | 67 | TGT | −63 | ||
| rrnL | 8842–9939 | 1098 | −41 | |||
| trnC | 9899–9964 | 66 | GCA | 1 | ||
| rrnS | 9966–10,687 | 722 | 29 | |||
| cox2 | 10,717–11,325 | 509 | GTG | TAA | 0 | |
| nad6 | 11,326–11,781 | 456 | ATG | TAA | −1 | |
| trnY | 11,781–11,853 | 73 | GTA | 7 | ||
| trnL1 | 11,861–11,926 | 66 | TAG | 10 | ||
| trnS2 | 11,937–11,996 | 60 | TGA | 2 | ||
| trnL2 | 11,999–12,062 | 64 | TAA | 29 | ||
| trnR | 12,092–12,155 | 64 | CGA | 3 | ||
| nad5 | 12,159–13,775 | 1617 | ATG | TAG | −10 | |
| trnG | 13,766–13,831 | 66 | TCC | 371 | ||
| SNCR | 13,832–14,202 | 371 | 0 | |||
| trnE | 14,203–14,267 | 65 | TTC | 0 | ||
| LNCR | 14,268–15,831 | 1564 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, F.; Li, X.; Ye, B.; Zhou, Y.; Dang, Z.; Tang, W.; Wang, L.; Zhang, H.; Chui, W.; Kui, J. Characterization of the Complete Mitochondrial Genome and Phylogenetic Analyses of Eurytrema coelomaticum (Trematoda: Dicrocoeliidae). Genes 2023, 14, 2199. https://doi.org/10.3390/genes14122199
Huang F, Li X, Ye B, Zhou Y, Dang Z, Tang W, Wang L, Zhang H, Chui W, Kui J. Characterization of the Complete Mitochondrial Genome and Phylogenetic Analyses of Eurytrema coelomaticum (Trematoda: Dicrocoeliidae). Genes. 2023; 14(12):2199. https://doi.org/10.3390/genes14122199
Chicago/Turabian StyleHuang, Fuqiang, Xin Li, Bijin Ye, Yule Zhou, Zhisheng Dang, Wenqiang Tang, Long Wang, Haoji Zhang, Wenting Chui, and Jun Kui. 2023. "Characterization of the Complete Mitochondrial Genome and Phylogenetic Analyses of Eurytrema coelomaticum (Trematoda: Dicrocoeliidae)" Genes 14, no. 12: 2199. https://doi.org/10.3390/genes14122199
APA StyleHuang, F., Li, X., Ye, B., Zhou, Y., Dang, Z., Tang, W., Wang, L., Zhang, H., Chui, W., & Kui, J. (2023). Characterization of the Complete Mitochondrial Genome and Phylogenetic Analyses of Eurytrema coelomaticum (Trematoda: Dicrocoeliidae). Genes, 14(12), 2199. https://doi.org/10.3390/genes14122199