
Mitochondria, a Key Target in Amyotrophic Lateral Sclerosis Pathogenesis


Abstract
1. Amyotrophic Lateral Sclerosis, a Motor Neuron Disease
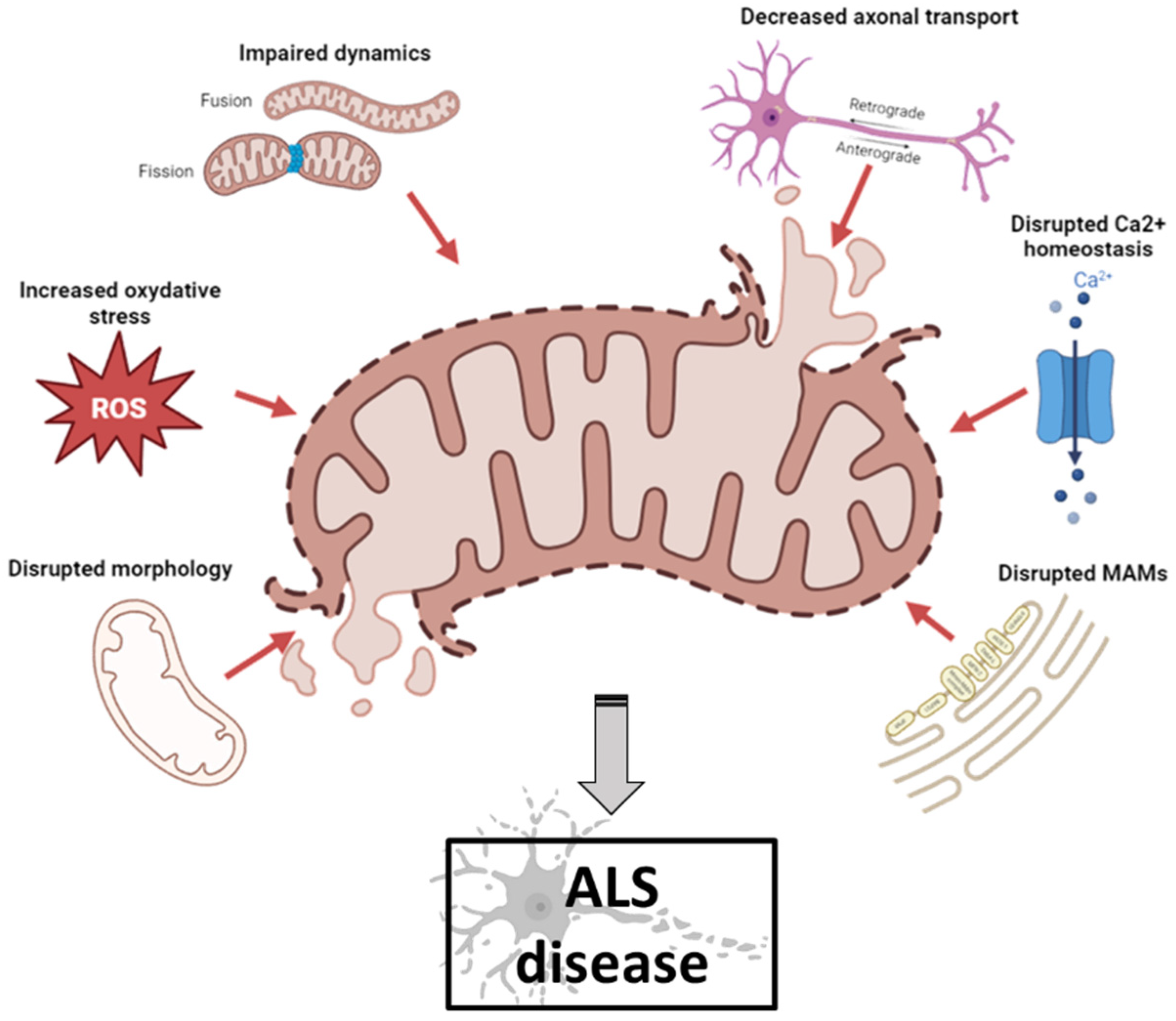
2. Mitochondrial Dysfunction in ALS Pathogenesis
2.1. Mitochondria and ALS
2.2. Mitochondria and Ultrastructural Morphology
2.3. Mitochondria and Reactive Oxidative Species (ROS)
2.4. Mitochondria and Dynamics
2.5. Mitochondria and Axonal Transport
2.6. Mitochondria and Calcium Homeostasis
2.7. Mitochondria and MAM
3. Secondary Mitochondrial Dysfunctions in ALS
3.1. SOD1/ALS1
3.2. C9ORF72/ALS-FTD1
3.3. TDP-43/ALS10
3.4. FUS/ALS6
3.5. Alsin/ALS2
3.6. VAPB/ALS8
3.7. OPTN/ALS12
3.8. SIGMAR1/ALS16
3.9. SQSTM1/ALS-FTD3
3.10. VCP/ALS14
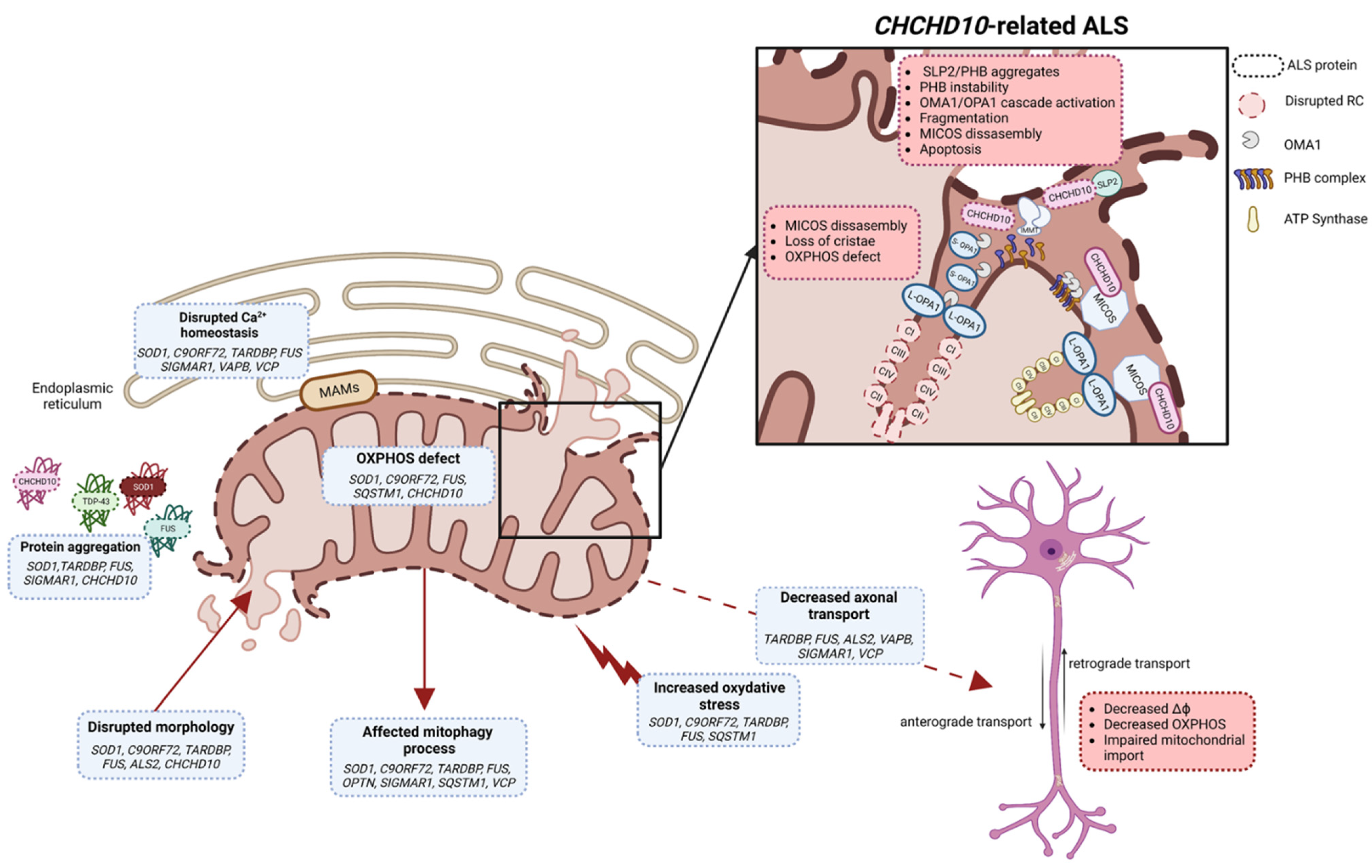
4. Primary Mitochondrial Dysfunction in CHCHD10-Related ALS
4.1. CHCHD10/ALS-FTD2
4.2. CHCHD10 and Mitochondrial Impairments in ALS/FTD
5. Current Drugs Used in ALS
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| fALS | familial ALS |
| sALS | sporadic ALS |
| CHCHD10 | Coiled-coil-Helix-Coiled-coil-Helix Domain containing 10 |
| DRP1 | dynamin-related protein 1 |
| ER | endoplasmic reticulum |
| FIS1 | fission protein 1 |
| FTD | frontotemporal dementia |
| FUS | fused in sarcoma |
| IMM | inner mitochondrial membrane |
| IMS | intermembrane space |
| IP3 | inositol 1,4,5-triphosphate |
| IP3R | inositol 1,4,5-triphosphate receptor |
| MAM | mitochondrial-associated membranes |
| MFN | mitofusin |
| MICOS | mitochondrial contact site and cristae organizing system |
| MN | motor neuron |
| MND | motor neuron disease |
| mtDNA | mitochondrial DNA |
| mt-ISR | mitochondrial integrated stress response |
| NMJ | neuromuscular junction |
| OMM | outer mitochondrial membrane |
| OPA1 | optic atrophy 1 |
| OPTN | optineurin |
| OXPHOS | oxidative phosphorylation |
| PHB | prohibitin |
| PTPIP51 | protein tyrosine phosphatase interacting protein-51 |
| ROS | reactive oxygen species |
| SLP2 | stomatin-like protein 2 |
| SOD1 | superoxide dismutase 1 |
| TARDBP/TDP-43 | trans-activating response region DNA-binding protein 43 |
| UPR | unfolded protein response |
| VAPB | vesicle-associated membrane protein-associated protein B |
| VCP | vasolin-containing protein |
| VDAC | voltage-dependent anion-selective channel proteins |
References
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; Van Den Berg, L.H. Amyotrophic Lateral Sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, T.; Liu, L.; Yao, X.; Chen, L.; Fan, D.; Zhan, S.; Wang, S. Global Variation in Prevalence and Incidence of Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. J. Neurol. 2020, 267, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Marin, B.; Boumédiene, F.; Logroscino, G.; Couratier, P.; Babron, M.-C.; Leutenegger, A.L.; Copetti, M.; Preux, P.-M.; Beghi, E. Variation in Worldwide Incidence of Amyotrophic Lateral Sclerosis: A Meta-Analysis. Int. J. Epidemiol. 2016, 46, dyw061. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C.; Goldstein, L.A.; White, L.A. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef]
- Ryan, M.; Heverin, M.; McLaughlin, R.L.; Hardiman, O. Lifetime Risk and Heritability of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2019, 76, 1367. [Google Scholar] [CrossRef] [PubMed]
- Logroscino, G.; Piccininni, M.; Marin, B.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Alahdab, F.; Asgedom, S.W.; Awasthi, A.; Chaiah, Y.; et al. Global, Regional, and National Burden of Motor Neuron Diseases 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Wijesekera, L.C.; Nigel Leigh, P. Amyotrophic Lateral Sclerosis. Orphanet J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef]
- Zufiría, M.; Gil-Bea, F.J.; Fernández-Torrón, R.; Poza, J.J.; Muñoz-Blanco, J.L.; Rojas-García, R.; Riancho, J.; López De Munain, A. ALS: A Bucket of Genes, Environment, Metabolism and Unknown Ingredients. Prog. Neurobiol. 2016, 142, 104–129. [Google Scholar] [CrossRef]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Emerging Insights into the Complex Genetics and Pathophysiology of Amyotrophic Lateral Sclerosis. Lancet Neurol. 2022, 21, 465–479. [Google Scholar] [CrossRef]
- Parobkova, E.; Matej, R. Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degenerations: Similarities in Genetic Background. Diagnostics 2021, 11, 509. [Google Scholar] [CrossRef]
- De Conti, L.; Borroni, B.; Baralle, M. New Routes in Frontotemporal Dementia Drug Discovery. Expert Opin. Drug Discov. 2017, 12, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M. Molecular Neuropathology of TDP-43 Proteinopathies. IJMS 2009, 10, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Rademakers, R.; Neumann, M. TDP-43 and FUS in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s Something Wrong with My MAM; the ER–Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef]
- Lau, D.H.W.; Hartopp, N.; Welsh, N.J.; Mueller, S.; Glennon, E.B.; Mórotz, G.M.; Annibali, A.; Gomez-Suaga, P.; Stoica, R.; Paillusson, S.; et al. Disruption of ER−mitochondria Signalling in Fronto-Temporal Dementia and Related Amyotrophic Lateral Sclerosis. Cell Death Dis. 2018, 9, 327. [Google Scholar] [CrossRef] [PubMed]
- Markovinovic, A.; Greig, J.; Martín-Guerrero, S.M.; Salam, S.; Paillusson, S. Endoplasmic Reticulum–Mitochondria Signaling in Neurons and Neurodegenerative Diseases. J. Cell Sci. 2022, 135, jcs248534. [Google Scholar] [CrossRef]
- Cozzolino, M.; Rossi, S.; Mirra, A.; Carrì, M.T. Mitochondrial Dynamism and the Pathogenesis of Amyotrophic Lateral Sclerosis. Front. Cell Neurosci. 2015, 9, 31. [Google Scholar] [CrossRef]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A Mitochondrial Origin for Frontotemporal Dementia and Amyotrophic Lateral Sclerosis through CHCHD10 Involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Glynn, S.M.; Gibbs, J.R.; Nalls, M.A.; Sabatelli, M.; Restagno, G.; Drory, V.E.; Chiò, A.; Rogaeva, E.; Traynor, B.J. Mutations in the CHCHD10 Gene Are a Common Cause of Familial Amyotrophic Lateral Sclerosis. Brain 2014, 137, e311. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Andersen, P.M.; Hübers, A.; Marroquin, N.; Volk, A.E.; Danzer, K.M.; Meitinger, T.; Ludolph, A.C.; Strom, T.M.; Weishaupt, J.H. Two Novel Mutations in Conserved Codons Indicate That CHCHD10 Is a Gene Associated with Motor Neuron Disease. Brain 2014, 137, e309. [Google Scholar] [CrossRef] [PubMed]
- Sirkis, D.W.; Geier, E.G.; Bonham, L.W.; Karch, C.M.; Yokoyama, J.S. Recent Advances in the Genetics of Frontotemporal Dementia. Curr. Genet. Med. Rep. 2019, 7, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Carrì, M.T. Mitochondrial Dysfunction in ALS. Prog. Neurobiol. 2012, 97, 54–66. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Mitochondrial Alterations in the Spinal Cord of Patients With Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef]
- Genin, E.C.; Madji Hounoum, B.; Bannwarth, S.; Fragaki, K.; Lacas-Gervais, S.; Mauri-Crouzet, A.; Lespinasse, F.; Neveu, J.; Ropert, B.; Augé, G.; et al. Mitochondrial Defect in Muscle Precedes Neuromuscular Junction Degeneration and Motor Neuron Death in CHCHD10S59L/+ Mouse. Acta Neuropathol. 2019, 138, 123–145. [Google Scholar] [CrossRef]
- Singh, T.; Jiao, Y.; Ferrando, L.M.; Yablonska, S.; Li, F.; Horoszko, E.C.; Lacomis, D.; Friedlander, R.M.; Carlisle, D.L. Neuronal Mitochondrial Dysfunction in Sporadic Amyotrophic Lateral Sclerosis Is Developmentally Regulated. Sci. Rep. 2021, 11, 18916. [Google Scholar] [CrossRef]
- Magrané, J.; Cortez, C.; Gan, W.-B.; Manfredi, G. Abnormal Mitochondrial Transport and Morphology Are Common Pathological Denominators in SOD1 and TDP43 ALS Mouse Models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Vercesi, A.E. Mitochondrial Damage Induced by Conditions of Oxidative Stress. Free Radic. Biol. Med. 1999, 26, 463–471. [Google Scholar] [CrossRef]
- Gandhi, S.; Abramov, A.Y. Mechanism of Oxidative Stress in Neurodegeneration. Oxidative Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Langer, T. Mitofusin 2 Builds a Bridge between ER and Mitochondria. Cell 2008, 135, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Yapa, N.M.B.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial Dynamics in Health and Disease. FEBS Lett. 2021, 595, 1184–1204. [Google Scholar] [CrossRef] [PubMed]
- Sebastián, D.; Palacín, M.; Zorzano, A. Mitochondrial Dynamics: Coupling Mitochondrial Fitness with Healthy Aging. Trends Mol. Med. 2017, 23, 201–215. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics-Fusion, Fission, Movement, and Mitophagy-in Neurodegenerative Diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.W.; Price, D.L. Axonal Transport in Motor Neuron Pathology. UCLA Forum Med. Sci. 1976, 19, 33–67. [Google Scholar]
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L.F. Axonal Transport: Cargo-Specific Mechanisms of Motility and Regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef]
- Sheng, Z.-H.; Cai, Q. Mitochondrial Transport in Neurons: Impact on Synaptic Homeostasis and Neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The Machineries, Regulation and Cellular Functions of Mitochondrial Calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-Associated Membranes: Composition, Molecular Mechanisms, and Physiopathological Implications. Antioxid. Redox Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and Endoplasmic Reticulum Calcium Homeostasis and Cell Death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Mak, D.-O.D.; Cheung, K.-H.; Toglia, P.; Foskett, J.K.; Ullah, G. Analyzing and Quantifying the Gain-of-Function Enhancement of IP3 Receptor Gating by Familial Alzheimer’s Disease-Causing Mutants in Presenilins. PLoS Comput. Biol. 2015, 11, e1004529. [Google Scholar] [CrossRef]
- Kania, E.; Roest, G.; Vervliet, T.; Parys, J.B.; Bultynck, G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 2017, 7, 140. [Google Scholar] [CrossRef]
- Valladares, D.; Utreras-Mendoza, Y.; Campos, C.; Morales, C.; Diaz-Vegas, A.; Contreras-Ferrat, A.; Westermeier, F.; Jaimovich, E.; Marchi, S.; Pinton, P.; et al. IP3 Receptor Blockade Restores Autophagy and Mitochondrial Function in Skeletal Muscle Fibers of Dystrophic Mice. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 3685–3695. [Google Scholar] [CrossRef]
- Honrath, B.; Matschke, L.; Meyer, T.; Magerhans, L.; Perocchi, F.; Ganjam, G.K.; Zischka, H.; Krasel, C.; Gerding, A.; Bakker, B.M.; et al. SK2 Channels Regulate Mitochondrial Respiration and Mitochondrial Ca2+ Uptake. Cell Death Differ. 2017, 24, 761–773. [Google Scholar] [CrossRef]
- Xu, H.; Guan, N.; Ren, Y.-L.; Wei, Q.-J.; Tao, Y.-H.; Yang, G.-S.; Liu, X.-Y.; Bu, D.-F.; Zhang, Y.; Zhu, S.-N. IP3R-Grp75-VDAC1-MCU Calcium Regulation Axis Antagonists Protect Podocytes from Apoptosis and Decrease Proteinuria in an Adriamycin Nephropathy Rat Model. BMC Nephrol. 2018, 19, 140. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Giorgi, C.; Bonora, M.; Punzetti, S.; Pavasini, R.; Wieckowski, M.R.; Campo, G.; Pinton, P. Molecular Identity of the Mitochondrial Permeability Transition Pore and Its Role in Ischemia-Reperfusion Injury. J. Mol. Cell. Cardiol. 2015, 78, 142–153. [Google Scholar] [CrossRef]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial Permeability Transition Involves Dissociation of F1FO ATP Synthase Dimers and C-ring Conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Fujimoto, M.; Hayashi, T. New Insights into the Role of Mitochondria-Associated Endoplasmic Reticulum Membrane. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2011; Volume 292, pp. 73–117. [Google Scholar]
- Area-Gomez, E.; Schon, E.A. On the Pathogenesis of Alzheimer’s Disease: The MAM Hypothesis. FASEB J. 2017, 31, 864–867. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Paillusson, S.; Miller, C.C.J. ER-Mitochondria Signaling Regulates Autophagy. Autophagy 2017, 13, 1250–1251. [Google Scholar] [CrossRef]
- Maity, S.; Komal, P.; Kumar, V.; Saxena, A.; Tungekar, A.; Chandrasekar, V. Impact of ER Stress and ER-Mitochondrial Crosstalk in Huntington’s Disease. IJMS 2022, 23, 780. [Google Scholar] [CrossRef]
- Cerqua, C.; Anesti, V.; Pyakurel, A.; Liu, D.; Naon, D.; Wiche, G.; Baffa, R.; Dimmer, K.S.; Scorrano, L. Trichoplein/Mitostatin Regulates Endoplasmic Reticulum–Mitochondria Juxtaposition. EMBO Rep. 2010, 11, 854–860. [Google Scholar] [CrossRef]
- Arasaki, K.; Shimizu, H.; Mogari, H.; Nishida, N.; Hirota, N.; Furuno, A.; Kudo, Y.; Baba, M.; Baba, N.; Cheng, J.; et al. A Role for the Ancient SNARE Syntaxin 17 in Regulating Mitochondrial Division. Dev. Cell 2015, 32, 304–317. [Google Scholar] [CrossRef]
- Iwasawa, R.; Mahul-Mellier, A.-L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 Bridge the Mitochondria-ER Interface to Establish a Platform for Apoptosis Induction: Fis1 Induces Apoptosis via Bap31. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Watanabe, S.; Komine, O.; Sobue, A.; Yamanaka, K. Novel Reporters of Mitochondria-associated Membranes (MAM), MAMtrackers, Demonstrate MAM Disruption as a Common Pathological Feature in Amyotrophic Lateral Sclerosis. FASEB J. 2021, 35, e21688. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Bunton-Stasyshyn, R.K.A.; Saccon, R.A.; Fratta, P.; Fisher, E.M.C. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist 2015, 21, 519–529. [Google Scholar] [CrossRef]
- Bowling, A.C.; Schulz, J.B.; Brown, R.H.; Beal, M.F. Superoxide Dismutase Activity, Oxidative Damage, and Mitochondrial Energy Metabolism in Familial and Sporadic Amyotrophic Lateral Sclerosis. J. Neurochem. 1993, 61, 2322–2325. [Google Scholar] [CrossRef]
- Zhao, X.; Feng, X.; Li, X.; Mou, J.; Liu, H.; Chen, J.; Wu, J. The G41D Mutation in SOD1-Related Amyotrophic Lateral Sclerosis Exhibits Phenotypic Heterogeneity among Individuals: A Case Report and Literature Review. Medicine 2022, 101, e28771. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Rakhit, R.; Cunningham, P.; Furtos-Matei, A.; Dahan, S.; Qi, X.-F.; Crow, J.P.; Cashman, N.R.; Kondejewski, L.H.; Chakrabartty, A. Oxidation-Induced Misfolding and Aggregation of Superoxide Dismutase and Its Implications for Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2002, 277, 47551–47556. [Google Scholar] [CrossRef]
- Higgins, C.M.; Jung, C.; Xu, Z. ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003, 15, 16. [Google Scholar]
- Pickles, S.; Destroismaisons, L.; Peyrard, S.L.; Cadot, S.; Rouleau, G.A.; Brown, R.H.; Julien, J.-P.; Arbour, N.; Vande Velde, C. Mitochondrial Damage Revealed by Immunoselection for ALS-Linked Misfolded SOD1. Hum. Mol. Genet. 2013, 22, 3947–3959. [Google Scholar] [CrossRef] [PubMed]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded Mutant SOD1 Directly Inhibits VDAC1 Conductance in a Mouse Model of Inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef]
- Pedrini, S.; Sau, D.; Guareschi, S.; Bogush, M.; Brown, R.H.; Naniche, N.; Kia, A.; Trotti, D.; Pasinelli, P. ALS-Linked Mutant SOD1 Damages Mitochondria by Promoting Conformational Changes in Bcl-2. Hum. Mol. Genet. 2010, 19, 2974–2986. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated Membrane Collapse Is a Common Pathomechanism in SIGMAR 1- and SOD 1-linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef]
- Tak, Y.J.; Park, J.-H.; Rhim, H.; Kang, S. ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin. Int. J. Mol. Sci. 2020, 21, 7525. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Xiang, S.; Kato, M.; Wu, L.; Theodoropoulos, P.; Wang, T.; Kim, J.; Yun, J.; Xie, Y.; McKnight, S.L. Poly-Dipeptides Encoded by the C9orf72 Repeats Bind Nucleoli, Impede RNA Biogenesis, and Kill Cells. Science 2014, 345, 1139–1145. [Google Scholar] [CrossRef]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 Repeat Expansions Cause Neurodegeneration in Drosophila through Arginine-Rich Proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense Proline-Arginine RAN Dipeptides Linked to C9ORF72-ALS/FTD Form Toxic Nuclear Aggregates That Initiate In Vitro and In Vivo Neuronal Death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef]
- Dols-Icardo, O.; Garcia-Redondo, A.; Rojas-Garcia, R.; Sanchez-Valle, R.; Noguera, A.; Gomez-Tortosa, E.; Pastor, P.; Hernandez, I.; Esteban-Perez, J.; Suarez-Calvet, M.; et al. Characterization of the Repeat Expansion Size in C9orf72 in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Hum. Mol. Genet. 2014, 23, 749–754. [Google Scholar] [CrossRef]
- Smeyers, J.; Banchi, E.-G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell. Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef]
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Taylor, J.P. The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci. 2017, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.N.; Wu, Y.; Odeh, H.M.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Yue, M.; Jiang, P.; Gomes, E.; Tong, J.; et al. C9orf72 Poly(GR) Aggregation Induces TDP-43 Proteinopathy. Sci. Transl. Med. 2020, 12, eabb3774. [Google Scholar] [CrossRef]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-Specific Mitochondria Dysfunctions in Human TARDBP and C9ORF72 Fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, Z.; Tantray, I.; Li, Y.; Chen, S.; Dong, J.; Glynn, S.; Vogel, H.; Snyder, M.; Lu, B. Quality-Control Mechanisms Targeting Translationally Stalled and C-Terminally Extended Poly(GR) Associated with ALS/FTD. Proc. Natl. Acad. Sci. USA 2020, 117, 25104–25115. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.-B. Poly(GR) in C9ORF72 -Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Lopez-Gonzalez, R.; Krishnan, G.; Phillips, H.L.; Li, A.N.; Seeley, W.W.; Yao, W.-D.; Almeida, S.; Gao, F.-B. C9ORF72-ALS/FTD-Associated Poly(GR) Binds Atp5a1 and Compromises Mitochondrial Function in Vivo. Nat. Neurosci. 2019, 22, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.L.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 Regulates Energy Homeostasis by Stabilizing Mitochondrial Complex I Assembly. Cell Metab. 2021, 33, 531–546.e9. [Google Scholar] [CrossRef]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial Bioenergetic Deficits in C9orf72 Amyotrophic Lateral Sclerosis Motor Neurons Cause Dysfunctional Axonal Homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.-H.; Hung, S.-T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y.; et al. Haploinsufficiency Leads to Neurodegeneration in C9ORF72 ALS/FTD Human Induced Motor Neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef]
- Mis, M.S.C.; Brajkovic, S.; Tafuri, F.; Bresolin, N.; Comi, G.P.; Corti, S. Development of Therapeutics for C9ORF72 ALS/FTD-Related Disorders. Mol. Neurobiol. 2017, 54, 4466–4476. [Google Scholar] [CrossRef] [PubMed]
- Beckers, J.; Tharkeshwar, A.K.; Van Damme, P. C9orf72 ALS-FTD: Recent Evidence for Dysregulation of the Autophagy-Lysosome Pathway at Multiple Levels. Autophagy 2021, 17, 3306–3322. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, K.M.; Maulding, K.; Ruan, K.; Senturk, M.; Grima, J.C.; Sung, H.; Zuo, Z.; Song, H.; Gao, J.; Dubey, S.; et al. TFEB/Mitf Links Impaired Nuclear Import to Autophagolysosomal Dysfunction in C9-ALS. eLife 2020, 9, e59419. [Google Scholar] [CrossRef]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER–Mitochondria Associations Are Regulated by the VAPB–PTPIP51 Interaction and Are Disrupted by ALS/FTD-Associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; De Vos, K.J.; Shaw, C.E.; et al. ALS/FTD -associated FUS Activates GSK -3β to Disrupt the VAPB–PTPIP 51 Interaction and ER –Mitochondria Associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Lee, V.M.-Y.; Trojanowski, J.Q. TAR DNA-Binding Protein 43 in Neurodegenerative Disease. Nat. Rev. Neurol. 2010, 6, 211–220. [Google Scholar] [CrossRef]
- Tagawa, A.; Tan, C.-F.; Kikugawa, K.; Fukase, M.; Nakano, R.; Onodera, O.; Nishizawa, M.; Takahashi, H. Familial Amyotrophic Lateral Sclerosis: A SOD1-Unrelated Japanese Family of Bulbar Type with Bunina Bodies and Ubiquitin-Positive Skein-like Inclusions in Lower Motor Neurons. Acta Neuropathol. 2007, 113, 205–211. [Google Scholar] [CrossRef]
- Yokoseki, A.; Shiga, A.; Tan, C.-F.; Tagawa, A.; Kaneko, H.; Koyama, A.; Eguchi, H.; Tsujino, A.; Ikeuchi, T.; Kakita, A.; et al. TDP-43 Mutation in Familial Amyotrophic Lateral Sclerosis. Ann. Neurol. 2008, 63, 538–542. [Google Scholar] [CrossRef]
- Buratti, E.; Baralle, F.E. Characterization and Functional Implications of the RNA Binding Properties of Nuclear Factor TDP-43, a Novel Splicing Regulator ofCFTR Exon 9. J. Biol. Chem. 2001, 276, 36337–36343. [Google Scholar] [CrossRef] [PubMed]
- Pesiridis, G.S.; Lee, V.M.-Y.; Trojanowski, J.Q. Mutations in TDP-43 Link Glycine-Rich Domain Functions to Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2009, 18, R156–R162. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging Roles in RNA Processing and Neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E. Functional Significance of TDP-43 Mutations in Disease. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 2015; Volume 91, pp. 1–53. [Google Scholar]
- Benajiba, L.; Le Ber, I.; Camuzat, A.; Lacoste, M.; Thomas-Anterion, C.; Couratier, P.; Legallic, S.; Salachas, F.; Hannequin, D.; Decousus, M.; et al. TARDBP Mutations in Motoneuron Disease with Frontotemporal Lobar Degeneration. Ann. Neurol. 2009, 65, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.H.; Ke, Y.D.; Ittner, L.M.; Halliday, G.M. ALS/FTLD: Experimental Models and Reality. Acta Neuropathol. 2017, 133, 177–196. [Google Scholar] [CrossRef]
- Shan, X.; Chiang, P.-M.; Price, D.L.; Wong, P.C. Altered Distributions of Gemini of Coiled Bodies and Mitochondria in Motor Neurons of TDP-43 Transgenic Mice. Proc. Natl. Acad. Sci. USA 2010, 107, 16325–16330. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-F.; Gendron, T.F.; Zhang, Y.-J.; Lin, W.-L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-Type Human TDP-43 Expression Causes TDP-43 Phosphorylation, Mitochondrial Aggregation, Motor Deficits, and Early Mortality in Transgenic Mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Lin, W.-L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS Disease-Associated Mutant TDP-43 Impairs Mitochondrial Dynamics and Function in Motor Neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; Da Rocha, E.L.; et al. The Inhibition of TDP-43 Mitochondrial Localization Blocks Its Neuronal Toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 Interacts with Mitochondrial Proteins Critical for Mitophagy and Mitochondrial Dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef]
- Wang, W.; Arakawa, H.; Wang, L.; Okolo, O.; Siedlak, S.L.; Jiang, Y.; Gao, J.; Xie, F.; Petersen, R.B.; Wang, X. Motor-Coordinative and Cognitive Dysfunction Caused by Mutant TDP-43 Could Be Reversed by Inhibiting Its Mitochondrial Localization. Mol. Ther. 2017, 25, 127–139. [Google Scholar] [CrossRef]
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef]
- Khalil, B.; Liévens, J.-C. Mitochondrial Quality Control in Amyotrophic Lateral Sclerosis: Towards a Common Pathway? Neural Regen. Res. 2017, 12, 1052. [Google Scholar] [CrossRef] [PubMed]
- Izumikawa, K.; Nobe, Y.; Yoshikawa, H.; Ishikawa, H.; Miura, Y.; Nakayama, H.; Nonaka, T.; Hasegawa, M.; Egawa, N.; Inoue, H.; et al. TDP-43 Stabilises the Processing Intermediates of Mitochondrial Transcripts. Sci. Rep. 2017, 7, 7709. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Gao, K.; Jankovic, J. The Role of FUS Gene Variants in Neurodegenerative Diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.A.; Huang, E.J.; Tsai, L.-H. Interaction of FUS and HDAC1 Regulates DNA Damage Response and Repair in Neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Waibel, S.; Neumann, M.; Rosenbohm, A.; Birve, A.; Volk, A.E.; Weishaupt, J.H.; Meyer, T.; Müller, U.; Andersen, P.M.; Ludolph, A.C. Truncating Mutations in FUS/TLS Give Rise to a More Aggressive ALS -phenotype than Missense Mutations: A Clinico-genetic Study in Germany. Eur. J. Neurol. 2013, 20, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Baumer, D.; Hilton, D.; Paine, S.M.L.; Turner, M.R.; Lowe, J.; Talbot, K.; Ansorge, O. Juvenile ALS with Basophilic Inclusions Is a FUS Proteinopathy with FUS Mutations. Neurology 2010, 75, 611–618. [Google Scholar] [CrossRef]
- Gromicho, M.; Oliveira Santos, M.; Pinto, A.; Pronto-Laborinho, A.; De Carvalho, M. Young-Onset Rapidly Progressive ALS Associated with Heterozygous FUS Mutation. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-Y.; Cui, L.-Y.; Sun, Q.; Li, X.-G.; Liu, M.-S.; Xu, Y.; Zhou, Y.; Yang, X.-Z. De Novo FUS Gene Mutations Are Associated with Juvenile-Onset Sporadic Amyotrophic Lateral Sclerosis in China. Neurobiol. Aging 2013, 34, 1312.e1–1312.e8. [Google Scholar] [CrossRef]
- Van Langenhove, T.; Van Der Zee, J.; Sleegers, K.; Engelborghs, S.; Vandenberghe, R.; Gijselinck, I.; Van Den Broeck, M.; Mattheijssens, M.; Peeters, K.; De Deyn, P.P.; et al. Genetic Contribution of FUS to Frontotemporal Lobar Degeneration. Neurology 2010, 74, 366–371. [Google Scholar] [CrossRef]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobágyi, T.; et al. ALS Mutant FUS Disrupts Nuclear Localization and Sequesters Wild-Type FUS within Cytoplasmic Stress Granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef] [PubMed]
- Sasayama, H.; Shimamura, M.; Tokuda, T.; Azuma, Y.; Yoshida, T.; Mizuno, T.; Nakagawa, M.; Fujikake, N.; Nagai, Y.; Yamaguchi, M. Knockdown of the Drosophila Fused in Sarcoma (FUS) Homologue Causes Deficient Locomotive Behavior and Shortening of Motoneuron Terminal Branches. PLoS ONE 2012, 7, e39483. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.C.; McGoldrick, P.; Vance, C.; Hortobagyi, T.; Sreedharan, J.; Rogelj, B.; Tudor, E.L.; Smith, B.N.; Klasen, C.; Miller, C.C.J.; et al. Overexpression of Human Wild-Type FUS Causes Progressive Motor Neuron Degeneration in an Age- and Dose-Dependent Fashion. Acta Neuropathol. 2013, 125, 273–288. [Google Scholar] [CrossRef] [PubMed]
- So, E.; Mitchell, J.C.; Memmi, C.; Chennell, G.; Vizcay-Barrena, G.; Allison, L.; Shaw, C.E.; Vance, C. Mitochondrial Abnormalities and Disruption of the Neuromuscular Junction Precede the Clinical Phenotype and Motor Neuron Loss in hFUSWT Transgenic Mice. Hum. Mol. Genet. 2018, 27, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.S.; Tungtur, S.; Tanaka, T.; Nadeau, L.L.; Badawi, Y.; Wang, H.; Ni, H.-M.; Ding, W.-X.; Nishimune, H. Impaired Mitophagy Plays a Role in Denervation of Neuromuscular Junctions in ALS Mice. Front. Neurosci. 2017, 11, 473. [Google Scholar] [CrossRef]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.; et al. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet. 2015, 11, e1005357. [Google Scholar] [CrossRef]
- Salam, S.; Tacconelli, S.; Smith, B.N.; Mitchell, J.C.; Glennon, E.; Nikolaou, N.; Houart, C.; Vance, C. Identification of a Novel Interaction of FUS and Syntaphilin May Explain Synaptic and Mitochondrial Abnormalities Caused by ALS Mutations. Sci. Rep. 2021, 11, 13613. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C.; et al. Divergent Roles of ALS-Linked Proteins FUS/TLS and TDP-43 Intersect in Processing Long Pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Colombrita, C.; Onesto, E.; Megiorni, F.; Pizzuti, A.; Baralle, F.E.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 and FUS RNA-Binding Proteins Bind Distinct Sets of Cytoplasmic Messenger RNAs and Differently Regulate Their Post-Transcriptional Fate in Motoneuron-like Cells. J. Biol. Chem. 2012, 287, 15635–15647. [Google Scholar] [CrossRef]
- Tsai, Y.-L.; Coady, T.H.; Lu, L.; Zheng, D.; Alland, I.; Tian, B.; Shneider, N.A.; Manley, J.L. ALS/FTD-Associated Protein FUS Induces Mitochondrial Dysfunction by Preferentially Sequestering Respiratory Chain Complex mRNAs. Genes. Dev. 2020, 34, 785–805. [Google Scholar] [CrossRef]
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A Gene Encoding a Putative GTPase Regulator Is Mutated in Familial Amyotrophic Lateral Sclerosis 2. Nat. Genet. 2001, 29, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hentati, A.; Deng, H.-X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.-Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The Gene Encoding Alsin, a Protein with Three Guanine-Nucleotide Exchange Factor Domains, Is Mutated in a Form of Recessive Amyotrophic Lateral Sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Otomo, A.; Ueda, M.T.; Hiratsuka, Y.; Suzuki-Utsunomiya, K.; Sugiyama, J.; Murakoshi, S.; Mitsui, S.; Ono, S.; Nakagawa, S.; et al. Altered Oligomeric States in Pathogenic ALS2 Variants Associated with Juvenile Motor Neuron Diseases Cause Loss of ALS2-Mediated Endosomal Function. J. Biol. Chem. 2018, 293, 17135–17153. [Google Scholar] [CrossRef]
- Sprute, R.; Jergas, H.; Ölmez, A.; Alawbathani, S.; Karasoy, H.; Dafsari, H.S.; Becker, K.; Daimagüler, H.; Nürnberg, P.; Muntoni, F.; et al. Genotype–Phenotype Correlation in Seven Motor Neuron Disease Families with Novel ALS2 Mutations. Am. J. Med. Genet. Part A 2021, 185, 344–354. [Google Scholar] [CrossRef]
- Eymard-Pierre, E.; Lesca, G.; Dollet, S.; Santorelli, F.M.; Di Capua, M.; Bertini, E.; Boespflug-Tanguy, O. Infantile-Onset Ascending Hereditary Spastic Paralysis Is Associated with Mutations in the Alsin Gene. Am. J. Hum. Genet. 2002, 71, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Hamida, M.B.; Hentati, F.; Hamida, C.B. Hereditary motor system diseases (chronic juvenile amyotrophic lateral sclerosis): Conditions combining a bilateral pyramidal syndrome with limb and bulbar amyotrophy. Brain 1990, 113, 347–363. [Google Scholar] [CrossRef]
- Hentati, A.; Bejaoui, K.; Pericak-Vance, M.A.; Hentati, F.; Speer, M.C.; Hung, W.-Y.; Figlewicz, D.A.; Haines, J.; Rimmler, J.; Ben Hamida, C.; et al. Linkage of Recessive Familial Amyotrophic Lateral Sclerosis to Chromosome 2q33–Q35. Nat. Genet. 1994, 7, 425–428. [Google Scholar] [CrossRef]
- Otomo, A. ALS2, a Novel Guanine Nucleotide Exchange Factor for the Small GTPase Rab5, Is Implicated in Endosomal Dynamics. Hum. Mol. Genet. 2003, 12, 1671–1687. [Google Scholar] [CrossRef] [PubMed]
- Soares, D.C.; Barlow, P.N.; Porteous, D.J.; Devon, R.S. An Interrupted Beta-Propeller and Protein Disorder: Structural Bioinformatics Insights into the N-Terminus of Alsin. J. Mol. Model. 2009, 15, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Koçak Eker, H.; Ünlü, S.E.; Al-Salmi, F.; Crosby, A.H. A Novel Homozygous Mutation in ALS2 Gene in Four Siblings with Infantile-Onset Ascending Hereditary Spastic Paralysis. Eur. J. Med. Genet. 2014, 57, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Hadano, S.; Benn, S.C.; Kakuta, S.; Otomo, A.; Sudo, K.; Kunita, R.; Suzuki-Utsunomiya, K.; Mizumura, H.; Shefner, J.M.; Cox, G.A.; et al. Mice Deficient in the Rab5 Guanine Nucleotide Exchange Factor ALS2/Alsin Exhibit Age-Dependent Neurological Deficits and Altered Endosome Trafficking. Hum. Mol. Genet. 2006, 15, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Hadano, S.; Kunita, R.; Otomo, A.; Suzuki-Utsunomiya, K.; Ikeda, J.-E. Molecular and Cellular Function of ALS2/Alsin: Implication of Membrane Dynamics in Neuronal Development and Degeneration. Neurochem. Int. 2007, 51, 74–84. [Google Scholar] [CrossRef]
- Hsu, F.; Spannl, S.; Ferguson, C.; Hyman, A.A.; Parton, R.G.; Zerial, M. Rab5 and Alsin Regulate Stress-Activated Cytoprotective Signaling on Mitochondria. eLife 2018, 7, e32282. [Google Scholar] [CrossRef]
- Gautam, M.; Jara, J.H.; Sekerkova, G.; Yasvoina, M.V.; Martina, M.; Özdinler, P.H. Absence of Alsin Function Leads to Corticospinal Motor Neuron Vulnerability via Novel Disease Mechanisms. Hum. Mol. Genet. 2016, 25, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.A.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.M.; Gillingwater, T.; Webb, J.; et al. A Mutation in the Vesicle-Trafficking Protein VAPB Causes Late-Onset Spinal Muscular Atrophy and Amyotrophic Lateral Sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef]
- Kanekura, K.; Nishimoto, I.; Aiso, S.; Matsuoka, M. Characterization of Amyotrophic Lateral Sclerosis-Linked P56S Mutation of Vesicle-Associated Membrane Protein-Associated Protein B (VAPB/ALS8). J. Biol. Chem. 2006, 281, 30223–30233. [Google Scholar] [CrossRef] [PubMed]
- Teuling, E.; Ahmed, S.; Haasdijk, E.; Demmers, J.; Steinmetz, M.O.; Akhmanova, A.; Jaarsma, D.; Hoogenraad, C.C. Motor Neuron Disease-Associated Mutant Vesicle-Associated Membrane Protein-Associated Protein (VAP) B Recruits Wild-Type VAPs into Endoplasmic Reticulum-Derived Tubular Aggregates. J. Neurosci. 2007, 27, 9801–9815. [Google Scholar] [CrossRef]
- De Vos, K.J.; Mórotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.-F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C.J. VAPB Interacts with the Mitochondrial Protein PTPIP51 to Regulate Calcium Homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein Binds to the ER–Mitochondria Tethering Protein VAPB to Disrupt Ca2+ Homeostasis and Mitochondrial ATP Production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef]
- Gómez-Suaga, P.; Pérez-Nievas, B.G.; Glennon, E.B.; Lau, D.H.W.; Paillusson, S.; Mórotz, G.M.; Calì, T.; Pizzo, P.; Noble, W.; Miller, C.C.J. The VAPB-PTPIP51 Endoplasmic Reticulum-Mitochondria Tethering Proteins Are Present in Neuronal Synapses and Regulate Synaptic Activity. Acta Neuropathol. Commun. 2019, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Morotz, G.M.; De Vos, K.J.; Vagnoni, A.; Ackerley, S.; Shaw, C.E.; Miller, C.C.J. Amyotrophic Lateral Sclerosis-Associated Mutant VAPBP56S Perturbs Calcium Homeostasis to Disrupt Axonal Transport of Mitochondria. Hum. Mol. Genet. 2012, 21, 1979–1988. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Jang, A.; Reddy, R.; Yoon, W.H.; Jankowsky, J.L. Neuronal Overexpression of Human VAPB Slows Motor Impairment and Neuromuscular Denervation in a Mouse Model of ALS. Hum. Mol. Genet. 2016, 25, 4661–4673. [Google Scholar] [CrossRef]
- Toth, R.P.; Atkin, J.D. Dysfunction of Optineurin in Amyotrophic Lateral Sclerosis and Glaucoma. Front. Immunol. 2018, 9, 1017. [Google Scholar] [CrossRef]
- Li, Y.; Kang, J.; Horwitz, M.S. Interaction of an Adenovirus E3 14.7-Kilodalton Protein with a Novel Tumor Necrosis Factor Alpha-Inducible Cellular Protein Containing Leucine Zipper Domains. Mol. Cell. Biol. 1998, 18, 1601–1610. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of Optineurin in Amyotrophic Lateral Sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Pottier, C.; Bieniek, K.F.; Finch, N.; Van De Vorst, M.; Baker, M.; Perkersen, R.; Brown, P.; Ravenscroft, T.; Van Blitterswijk, M.; Nicholson, A.M.; et al. Whole-Genome Sequencing Reveals Important Role for TBK1 and OPTN Mutations in Frontotemporal Lobar Degeneration without Motor Neuron Disease. Acta Neuropathol. 2015, 130, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Che, C.; Feng, S.; Liu, C.; Li, L.; Li, Y.; Huang, H.; Zou, Z. Novel Mutation in Optineurin Causing Aggressive ALS+/−frontotemporal Dementia. Ann. Clin. Transl. Neurol. 2019, 6, 2377–2383. [Google Scholar] [CrossRef] [PubMed]
- Fifita, J.A.; Zhang, K.Y.; Galper, J.; Williams, K.L.; McCann, E.P.; Hogan, A.L.; Saunders, N.; Bauer, D.; Tarr, I.S.; Pamphlett, R.; et al. Genetic and Pathological Assessment of hnRNPA1, hnRNPA2/B1, and hnRNPA3 in Familial and Sporadic Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2017, 17, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Holzbaur, E.L.F. Spatiotemporal Dynamics of Autophagy Receptors in Selective Mitophagy. Autophagy 2016, 12, 1956–1957. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin Is an Autophagy Receptor for Damaged Mitochondria in Parkin-Mediated Mitophagy That Is Disrupted by an ALS-Linked Mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 Enhances Its Binding to Ub Chains and Promotes Selective Autophagy of Damaged Mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 Controls Autophagosomal Engulfment of Polyubiquitinated Mitochondria through P62/SQSTM1 Phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Su, T.-P. The Pharmacology of Sigma-1 Receptors. Pharmacol. Ther. 2009, 124, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Mavlyutov, T.A.; Epstein, M.L.; Andersen, K.A.; Ziskind-Conhaim, L.; Ruoho, A.E. The Sigma-1 Receptor Is Enriched in Postsynaptic Sites of C-Terminals in Mouse Motoneurons. An Anatomical and Behavioral Study. Neuroscience 2010, 167, 247–255. [Google Scholar] [CrossRef]
- Luty, A.A.; Kwok, J.B.J.; Dobson-Stone, C.; Loy, C.T.; Coupland, K.G.; Karlström, H.; Sobow, T.; Tchorzewska, J.; Maruszak, A.; Barcikowska, M.; et al. Sigma Nonopioid Intracellular Receptor 1 Mutations Cause Frontotemporal Lobar Degeneration-Motor Neuron Disease. Ann. Neurol. 2010, 68, 639–649. [Google Scholar] [CrossRef]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A Mutation in Sigma-1 Receptor Causes Juvenile Amyotrophic Lateral Sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Marissal, N.; Médard, J.-J.; Azzedine, H.; Chrast, R. Dysfunction in Endoplasmic Reticulum-Mitochondria Crosstalk Underlies SIGMAR1 Loss of Function Mediated Motor Neuron Degeneration. Brain 2015, 138, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Fecto, F. SQSTM1 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Arch. Neurol. 2011, 68, 1440. [Google Scholar] [CrossRef] [PubMed]
- Rubino, E.; Rainero, I.; Chio, A.; Rogaeva, E.; Galimberti, D.; Fenoglio, P.; Grinberg, Y.; Isaia, G.; Calvo, A.; Gentile, S.; et al. SQSTM1 Mutations in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Neurology 2012, 79, 1556–1562. [Google Scholar] [CrossRef]
- Hirano, M.; Nakamura, Y.; Saigoh, K.; Sakamoto, H.; Ueno, S.; Isono, C.; Miyamoto, K.; Akamatsu, M.; Mitsui, Y.; Kusunoki, S. Mutations in the Gene Encoding P62 in Japanese Patients with Amyotrophic Lateral Sclerosis. Neurology 2013, 80, 458–463. [Google Scholar] [CrossRef]
- Teyssou, E.; Takeda, T.; Lebon, V.; Boillée, S.; Doukouré, B.; Bataillon, G.; Sazdovitch, V.; Cazeneuve, C.; Meininger, V.; LeGuern, E.; et al. Mutations in SQSTM1 Encoding P62 in Amyotrophic Lateral Sclerosis: Genetics and Neuropathology. Acta Neuropathol. 2013, 125, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Toyoshima, Y.; Shiga, A.; Yokoseki, A.; Arakawa, K.; Sekine, Y.; Shimohata, T.; Ikeuchi, T.; Nishizawa, M.; Kakita, A.; et al. Sporadic ALS with Compound Heterozygous Mutations in the SQSTM1 Gene. Acta Neuropathol. 2013, 126, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Le Ber, I. SQSTM1 Mutations in French Patients With Frontotemporal Dementia or Frontotemporal Dementia With Amyotrophic Lateral Sclerosis. JAMA Neurol. 2013, 70, 1403–1410. [Google Scholar]
- Gennari, L.; Gianfrancesco, F.; Di Stefano, M.; Rendina, D.; Merlotti, D.; Esposito, T.; Gallone, S.; Fusco, P.; Rainero, I.; Fenoglio, P.; et al. SQSTM1 Gene Analysis and Gene-Environment Interaction in Paget’s Disease of Bone. J. Bone Min. Res. 2010, 25, 1375–1384. [Google Scholar] [CrossRef]
- Zatloukal, K.; Stumptner, C.; Fuchsbichler, A.; Heid, H.; Schnoelzer, M.; Kenner, L.; Kleinert, R.; Prinz, M.; Aguzzi, A.; Denk, H. P62 Is a Common Component of Cytoplasmic Inclusions in Protein Aggregation Diseases. Am. J. Pathol. 2002, 160, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Al-Sarraj, S.; King, A.; Troakes, C.; Smith, B.; Maekawa, S.; Bodi, I.; Rogelj, B.; Al-Chalabi, A.; Hortobágyi, T.; Shaw, C.E. P62 Positive, TDP-43 Negative, Neuronal Cytoplasmic and Intranuclear Inclusions in the Cerebellum and Hippocampus Define the Pathology of C9orf72-Linked FTLD and MND/ALS. Acta Neuropathol. 2011, 122, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Seibenhener, M.L.; Du, Y.; Diaz-Meco, M.-T.; Moscat, J.; Wooten, M.C.; Wooten, M.W. A Role for Sequestosome 1/P62 in Mitochondrial Dynamics, Import and Genome Integrity. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wooten, M.C.; Wooten, M.W. Oxidative Damage to the Promoter Region of SQSTM1/P62 Is Common to Neurodegenerative Disease. Neurobiol. Dis. 2009, 35, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial Fragmentation in Neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef]
- Bartolome, F.; Esteras, N.; Martin-Requero, A.; Boutoleau-Bretonniere, C.; Vercelletto, M.; Gabelle, A.; Le Ber, I.; Honda, T.; Dinkova-Kostova, A.T.; Hardy, J.; et al. Pathogenic P62/SQSTM1 Mutations Impair Energy Metabolism through Limitation of Mitochondrial Substrates. Sci. Rep. 2017, 7, 1666. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Saisho, K.; Shimanuki, M.; Nakada, K.; Shitara, H.; Sou, Y.; Kimura, M.; Sato, S.; Hattori, N.; Komatsu, M.; et al. P62/SQSTM1 Cooperates with Parkin for Perinuclear Clustering of Depolarized Mitochondria: Parkin and P62 for Mitochondrial Clustering. Genes. Cells 2010, 15, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Goode, A.; Butler, K.; Long, J.; Cavey, J.; Scott, D.; Shaw, B.; Sollenberger, J.; Gell, C.; Johansen, T.; Oldham, N.J.; et al. Defective Recognition of LC3B by Mutant SQSTM1/P62 Implicates Impairment of Autophagy as a Pathogenic Mechanism in ALS-FTLD. Autophagy 2016, 12, 1094–1104. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Nagano, Y.; Taylor, J.P.; Lim, K.L.; Yao, T.-P. Disease-Causing Mutations in Parkin Impair Mitochondrial Ubiquitination, Aggregation, and HDAC6-Dependent Mitophagy. J. Cell Biol. 2010, 189, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-Mediated Mitophagy Is Dependent on VDAC1 and P62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Koppers, M.; Van Blitterswijk, M.M.; Vlam, L.; Rowicka, P.A.; Van Vught, P.W.J.; Groen, E.J.N.; Spliet, W.G.M.; Engelen-Lee, J.; Schelhaas, H.J.; De Visser, M.; et al. VCP Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2012, 33, 837.e7–837.e13. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.-S.; Fuentealba, R.A.; Miller, S.E.; Jackson, E.; Piwnica-Worms, D.; Baloh, R.H.; Weihl, C.C. Valosin-Containing Protein (VCP) Is Required for Autophagy and Is Disrupted in VCP Disease. J. Cell Biol. 2009, 187, 875–888. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef]
- Van De Warrenburg, B.P.; Schouten, M.I.; De Bot, S.T.; Vermeer, S.; Meijer, R.; Pennings, M.; Gilissen, C.; Willemsen, M.A.; Scheffer, H.; Kamsteeg, E.-J. Clinical Exome Sequencing for Cerebellar Ataxia and Spastic Paraplegia Uncovers Novel Gene–Disease Associations and Unanticipated Rare Disorders. Eur. J. Hum. Genet. 2016, 24, 1460–1466. [Google Scholar] [CrossRef]
- Gonzalez, M.A.; Feely, S.M.; Speziani, F.; Strickland, A.V.; Danzi, M.; Bacon, C.; Lee, Y.; Chou, T.-F.; Blanton, S.H.; Weihl, C.C.; et al. A Novel Mutation in VCP Causes Charcot–Marie–Tooth Type 2 Disease. Brain 2014, 137, 2897–2902. [Google Scholar] [CrossRef]
- Kimonis, V.E.; Mehta, S.G.; Fulchiero, E.C.; Thomasova, D.; Pasquali, M.; Boycott, K.; Neilan, E.G.; Kartashov, A.; Forman, M.S.; Tucker, S.; et al. Clinical Studies in Familial VCP Myopathy Associated with Paget Disease of Bone and Frontotemporal Dementia. Am. J. Med. Genet. Part A 2008, 146, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.S.L.; Rademakers, R.; Miller, B.L. Frontotemporal Dementia: A Bridge between Dementia and Neuromuscular Disease. Ann. N. Y. Acad. Sci. 2015, 1338, 71–93. [Google Scholar] [CrossRef] [PubMed]
- Van Der Zee, J.; Pirici, D.; Van Langenhove, T.; Engelborghs, S.; Vandenberghe, R.; Hoffmann, M.; Pusswald, G.; Van Den Broeck, M.; Peeters, K.; Mattheijssens, M.; et al. Clinical Heterogeneity in 3 Unrelated Families Linked to VCP p.Arg159His. Neurology 2009, 73, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Qiu, H. Valosin-Containing Protein, a Calcium-Associated ATPase Protein, in Endoplasmic Reticulum and Mitochondrial Function and Its Implications for Diseases. Int. J. Mol. Sci. 2020, 21, 3842. [Google Scholar] [CrossRef]
- Weihl, C.C.; Pestronk, A.; Kimonis, V.E. Valosin-Containing Protein Disease: Inclusion Body Myopathy with Paget’s Disease of the Bone and Fronto-Temporal Dementia. Neuromuscul. Disord. 2009, 19, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Weihl, C.C. The VCP/P97 System at a Glance: Connecting Cellular Function to Disease Pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef]
- Yeo, B.K.; Hong, C.J.; Chung, K.M.; Woo, H.; Kim, K.; Jung, S.; Kim, E.-K.; Yu, S.-W. Valosin-Containing Protein Is a Key Mediator between Autophagic Cell Death and Apoptosis in Adult Hippocampal Neural Stem Cells Following Insulin Withdrawal. Mol. Brain 2016, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.; Avci, D.; Queisser, M.A.; Gutschmidt, A.; Dreher, L.-S.; Fenech, E.J.; Volkmar, N.; Hayashi, Y.; Hoppe, T.; Christianson, J.C. Conserved Cytoplasmic Domains Promote Hrd1 Ubiquitin Ligase Complex Formation for ER-Associated Degradation (ERAD). J. Cell Sci. 2017, 130, 3322–3335. [Google Scholar] [CrossRef]
- Joshi, V.; Upadhyay, A.; Kumar, A.; Mishra, A. Gp78 E3 Ubiquitin Ligase: Essential Functions and Contributions in Proteostasis. Front. Cell. Neurosci. 2017, 11, 259. [Google Scholar] [PubMed]
- Guo, X.; Sun, X.; Hu, D.; Wang, Y.-J.; Fujioka, H.; Vyas, R.; Chakrapani, S.; Joshi, A.U.; Luo, Y.; Mochly-Rosen, D.; et al. VCP Recruitment to Mitochondria Causes Mitophagy Impairment and Neurodegeneration in Models of Huntington’s Disease. Nat. Commun. 2016, 7, 12646. [Google Scholar] [PubMed]
- Lizano, P.; Rashed, E.; Stoll, S.; Zhou, N.; Wen, H.; Hays, T.T.; Qin, G.; Xie, L.-H.; Depre, C.; Qiu, H. The Valosin-Containing Protein Is a Novel Mediator of Mitochondrial Respiration and Cell Survival in the Heart in Vivo. Sci. Rep. 2017, 7, 46324. [Google Scholar]
- Fang, L.; Hemion, C.; Pinho Ferreira Bento, A.C.; Bippes, C.C.; Flammer, J.; Neutzner, A. Mitochondrial Function in Neuronal Cells Depends on P97/VCP/Cdc48-Mediated Quality Control. Front. Cell. Neurosci. 2015, 9, 16. [Google Scholar]
- Kim, N.C.; Tresse, E.; Kolaitis, R.-M.; Molliex, A.; Thomas, R.E.; Alami, N.H.; Wang, B.; Joshi, A.; Smith, R.B.; Ritson, G.P.; et al. VCP Is Essential for Mitochondrial Quality Control by PINK1/Parkin and This Function Is Impaired by VCP Mutations. Neuron 2013, 78, 65–80. [Google Scholar]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Tokatlidis, K. The Coiled Coil-helix-coiled Coil-helix Proteins May Be Redox Proteins. FEBS Lett. 2009, 583, 1699–1702. [Google Scholar] [PubMed]
- Chiò, A.; Mora, G.; Sabatelli, M.; Caponnetto, C.; Traynor, B.J.; Johnson, J.O.; Nalls, M.A.; Calvo, A.; Moglia, C.; Borghero, G.; et al. CHCH10 Mutations in an Italian Cohort of Familial and Sporadic Amyotrophic Lateral Sclerosis Patients. Neurobiol. Aging 2015, 36, 1767.e3–1767.e6. [Google Scholar] [PubMed]
- Lehmer, C.; Schludi, M.H.; Ransom, L.; Greiling, J.; Junghänel, M.; Exner, N.; Riemenschneider, H.; Van Der Zee, J.; Van Broeckhoven, C.; Weydt, P.; et al. A Novel CHCHD10 Mutation Implicates a Mia40-dependent Mitochondrial Import Deficit in ALS. EMBO Mol. Med. 2018, 10, e8558. [Google Scholar] [PubMed]
- Ronchi, D.; Riboldi, G.; Del Bo, R.; Ticozzi, N.; Scarlato, M.; Galimberti, D.; Corti, S.; Silani, V.; Bresolin, N.; Comi, G.P. CHCHD10 Mutations in Italian Patients with Sporadic Amyotrophic Lateral Sclerosis: Figure 1. Brain 2015, 138, e372. [Google Scholar] [CrossRef]
- Zhang, M.; Xi, Z.; Zinman, L.; Bruni, A.C.; Maletta, R.G.; Curcio, S.A.M.; Rainero, I.; Rubino, E.; Pinessi, L.; Nacmias, B.; et al. Mutation Analysis of CHCHD10 in Different Neurodegenerative Diseases. Brain 2015, 138, e380. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, Y.; Wei, Q.; Cao, B.; Wu, Y.; Zhao, B.; Ou, R.; Yang, J.; Chen, X.; Hadano, S.; et al. Mutation Screening of the CHCHD10 Gene in Chinese Patients with Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2017, 54, 3189–3194. [Google Scholar]
- Shen, S.; He, J.; Tang, L.; Zhang, N.; Fan, D. CHCHD10 Mutations in Patients with Amyotrophic Lateral Sclerosis in Mainland China. Neurobiol. Aging 2017, 54, 214.e7–214.e10. [Google Scholar]
- Teyssou, E.; Chartier, L.; Albert, M.; Bouscary, A.; Antoine, J.-C.; Camdessanché, J.-P.; Rotolo, F.; Couratier, P.; Salachas, F.; Seilhean, D.; et al. Genetic Analysis of CHCHD10 in French Familial Amyotrophic Lateral Sclerosis Patients. Neurobiol. Aging 2016, 42, 218.e1–218.e3. [Google Scholar] [PubMed]
- Dols-Icardo, O.; Nebot, I.; Gorostidi, A.; Ortega-Cubero, S.; Hernández, I.; Rojas-García, R.; García-Redondo, A.; Povedano, M.; Lladó, A.; Álvarez, V.; et al. Analysis of the CHCHD10 Gene in Patients with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis from Spain. Brain 2015, 138, e400. [Google Scholar]
- Chaussenot, A.; Le Ber, I.; Ait-El-Mkadem, S.; Camuzat, A.; De Septenville, A.; Bannwarth, S.; Genin, E.C.; Serre, V.; Augé, G.; Brice, A.; et al. Screening of CHCHD10 in a French Cohort Confirms the Involvement of This Gene in Frontotemporal Dementia with Amyotrophic Lateral Sclerosis Patients. Neurobiol. Aging 2014, 35, 2884.e1–2884.e4. [Google Scholar]
- Che, X.-Q.; Zhao, Q.-H.; Huang, Y.; Li, X.; Ren, R.-J.; Chen, S.-D.; Wang, G.; Guo, Q.-H. Genetic Features of MAPT, GRN, C9orf72 and CHCHD10 Gene Mutations in Chinese Patients with Frontotemporal Dementia. CAR 2017, 14, 1102–1108. [Google Scholar]
- Jiao, B.; Xiao, T.; Hou, L.; Gu, X.; Zhou, Y.; Zhou, L.; Tang, B.; Xu, J.; Shen, L. High Prevalence of CHCHD10 Mutation in Patients with Frontotemporal Dementia from China. Brain 2016, 139, e21. [Google Scholar] [CrossRef] [PubMed]
- Perrone, F.; Nguyen, H.P.; Van Mossevelde, S.; Moisse, M.; Sieben, A.; Santens, P.; De Bleecker, J.; Vandenbulcke, M.; Engelborghs, S.; Baets, J.; et al. Investigating the Role of ALS Genes CHCHD10 and TUBA4A in Belgian FTD-ALS Spectrum Patients. Neurobiol. Aging 2017, 51, 177.e9–177.e16. [Google Scholar]
- Auranen, M.; Ylikallio, E.; Shcherbii, M.; Paetau, A.; Kiuru-Enari, S.; Toppila, J.P.; Tyynismaa, H. CHCHD10 Variant p.(Gly66Val) Causes Axonal Charcot-Marie-Tooth Disease. Neurol. Genet. 2015, 1, e1. [Google Scholar]
- Pasanen, P.; Myllykangas, L.; Pöyhönen, M.; Kiuru-Enari, S.; Tienari, P.J.; Laaksovirta, H.; Toppila, J.; Ylikallio, E.; Tyynismaa, H.; Auranen, M. Intrafamilial Clinical Variability in Individuals Carrying the CHCHD10 Mutation Gly66Val. Acta Neurol. Scand. 2016, 133, 361–366. [Google Scholar] [PubMed]
- Penttilä, S.; Jokela, M.; Bouquin, H.; Saukkonen, A.M.; Toivanen, J.; Udd, B. Late Onset Spinal Motor Neuronopathy Is Caused by Mutation in CHCHD10. Ann. Neurol. 2015, 77, 163–172. [Google Scholar] [PubMed]
- Penttilä, S.; Jokela, M.; Saukkonen, A.M.; Toivanen, J.; Palmio, J.; Lähdesmäki, J.; Sandell, S.; Shcherbii, M.; Auranen, M.; Ylikallio, E.; et al. CHCHD10 Mutations and Motor Neuron Disease: The Distribution in Finnish Patients. J. Neurol. Neurosurg. Psychiatry 2017, 88, 272–277. [Google Scholar] [CrossRef]
- Jokela, M.E.; Joutsa, J.; Udd, B. Evolving Neuromuscular Phenotype in a Patient with a Heterozygous CHCHD10 p.G66V Mutation. J. Neurol. 2016, 263, 1461–1462. [Google Scholar] [CrossRef] [PubMed]
- Ajroud-Driss, S.; Fecto, F.; Ajroud, K.; Lalani, I.; Calvo, S.E.; Mootha, V.K.; Deng, H.-X.; Siddique, N.; Tahmoush, A.J.; Heiman-Patterson, T.D.; et al. Mutation in the Novel Nuclear-Encoded Mitochondrial Protein CHCHD10 in a Family with Autosomal Dominant Mitochondrial Myopathy. Neurogenetics 2015, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rubino, E.; Zhang, M.; Mongini, T.; Boschi, S.; Vercelli, L.; Vacca, A.; Govone, F.; Gai, A.; Giordana, M.T.; Grinberg, M.; et al. Mutation Analysis of CHCHD2 and CHCHD10 in Italian Patients with Mitochondrial Myopathy. Neurobiol. Aging 2018, 66, 181.e1–181.e2. [Google Scholar] [CrossRef]
- Shammas, M.K.; Huang, X.; Wu, B.P.; Fessler, E.; Song, I.Y.; Randolph, N.P.; Li, Y.; Bleck, C.K.; Springer, D.A.; Fratter, C.; et al. OMA1 Mediates Local and Global Stress Responses against Protein Misfolding in CHCHD10 Mitochondrial Myopathy. J. Clin. Invest. 2022, 132, e157504. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; Choe, Y.-J.; Bannwarth, S.; Kim, J.; Maitra, S.; Dorn, G.W.; Taylor, J.P.; Paquis-Flucklinger, V.; Kim, N.C. TDP-43 and PINK1 Mediate CHCHD10S59L Mutation-Induced Defects in Drosophila and in Vitro. Nat. Commun. 2021, 12, 1924. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Bredvik, K.; Burstein, S.R.; Davis, C.; Meadows, S.M.; Dash, J.; Case, L.; Milner, T.A.; Kawamata, H.; Zuberi, A.; et al. ALS/FTD Mutant CHCHD10 Mice Reveal a Tissue-Specific Toxic Gain-of-Function and Mitochondrial Stress Response. Acta Neuropathol. 2019, 138, 103–121. [Google Scholar] [CrossRef]
- Kurzwelly, D.; Krüger, S.; Biskup, S.; Heneka, M.T. A Distinct Clinical Phenotype in a German Kindred with Motor Neuron Disease Carrying a CHCHD10 Mutation. Brain 2015, 138, e376. [Google Scholar] [CrossRef]
- Heiman-Patterson, T.D.; Argov, Z.; Chavin, J.M.; Kalman, B.; Alder, H.; DiMauro, S.; Bank, W.; Tahmoush, A.J. Biochemical and Genetic Studies in a Family with Mitochondrial Myopathy. Muscle Nerve 1997, 20, 1219–1224. [Google Scholar] [CrossRef]
- Genin, E.C.; Plutino, M.; Bannwarth, S.; Villa, E.; Cisneros-Barroso, E.; Roy, M.; Ortega-Vila, B.; Fragaki, K.; Lespinasse, F.; Pinero-Martos, E.; et al. CHCHD10 Mutations Promote Loss of Mitochondrial Cristae Junctions with Impaired Mitochondrial Genome Maintenance and Inhibition of Apoptosis. EMBO Mol. Med. 2016, 8, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.C.; Bannwarth, S.; Ropert, B.; Lespinasse, F.; Mauri-Crouzet, A.; Augé, G.; Fragaki, K.; Cochaud, C.; Donnarumma, E.; Lacas-Gervais, S.; et al. CHCHD10 and SLP2 Control the Stability of the PHB Complex: A Key Factor for Motor Neuron Viability. Brain 2022, 145, 3415–3430. [Google Scholar] [CrossRef]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.-C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 Processing and Mitochondrial Fusion by M-AAA Protease Isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible Proteolytic Inactivation of OPA1 Mediated by the OMA1 Protease in Mammalian Cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of Mitochondrial Morphology through Proteolytic Cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef] [PubMed]
- Sayles, N.M.; Southwell, N.; McAvoy, K.; Kim, K.; Pesini, A.; Anderson, C.J.; Quinzii, C.; Cloonan, S.; Kawamata, H.; Manfredi, G. Mutant CHCHD10 Causes an Extensive Metabolic Rewiring That Precedes OXPHOS Dysfunction in a Murine Model of Mitochondrial Cardiomyopathy. Cell Rep. 2022, 38, 110475. [Google Scholar] [CrossRef]
- Burstein, S.R.; Valsecchi, F.; Kawamata, H.; Bourens, M.; Zeng, R.; Zuberi, A.; Milner, T.A.; Cloonan, S.M.; Lutz, C.; Barrientos, A.; et al. In Vitro and in Vivo Studies of the ALS-FTLD Protein CHCHD10 Reveal Novel Mitochondrial Topology and Protein Interactions. Hum. Mol. Genet. 2018, 27, 160–177. [Google Scholar] [CrossRef]
- Straub, I.R.; Janer, A.; Weraarpachai, W.; Zinman, L.; Robertson, J.; Rogaeva, E.; Shoubridge, E.A. Loss of CHCHD10–CHCHD2 Complexes Required for Respiration Underlies the Pathogenicity of a CHCHD10 Mutation in ALS. Hum. Mol. Genet. 2018, 27, 178–189. [Google Scholar] [CrossRef]
- Funayama, M.; Ohe, K.; Amo, T.; Furuya, N.; Yamaguchi, J.; Saiki, S.; Li, Y.; Ogaki, K.; Ando, M.; Yoshino, H.; et al. CHCHD2 Mutations in Autosomal Dominant Late-Onset Parkinson’s Disease: A Genome-Wide Linkage and Sequencing Study. Lancet Neurol. 2015, 14, 274–282. [Google Scholar] [CrossRef]
- Shi, C.; Mao, C.; Zhang, S.; Yang, J.; Song, B.; Wu, P.; Zuo, C.; Liu, Y.; Ji, Y.; Yang, Z.; et al. CHCHD2 Gene Mutations in Familial and Sporadic Parkinson’s Disease. Neurobiol. Aging 2016, 38, 217.e9–217.e13. [Google Scholar] [CrossRef]
- Che, X.-Q.; Zhao, Q.-H.; Huang, Y.; Li, X.; Ren, R.-J.; Chen, S.-D.; Guo, Q.-H.; Wang, G. Mutation Screening of the CHCHD2 Gene for Alzheimer’s Disease and Frontotemporal Dementia in Chinese Mainland Population. JAD 2018, 61, 1283–1288. [Google Scholar] [CrossRef]
- Ogaki, K.; Koga, S.; Heckman, M.G.; Fiesel, F.C.; Ando, M.; Labbé, C.; Lorenzo-Betancor, O.; Moussaud-Lamodière, E.L.; Soto-Ortolaza, A.I.; Walton, R.L.; et al. Mitochondrial Targeting Sequence Variants of the CHCHD2 Gene Are a Risk for Lewy Body Disorders. Neurology 2015, 85, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wu, B.P.; Nguyen, D.; Liu, Y.-T.; Marani, M.; Hench, J.; Bénit, P.; Kozjak-Pavlovic, V.; Rustin, P.; Frank, S.; et al. CHCHD2 Accumulates in Distressed Mitochondria and Facilitates Oligomerization of CHCHD10. Hum. Mol. Genet. 2018, 27, 3881–3900. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Wang, H.; Luo, H.; Zhang, S.; Xu, H.; Zhang, S.; Rosenblum, J.; Wang, Z.; Zhang, Q.; Tang, M.; et al. CHCHD10 Is Involved in the Development of Parkinson’s Disease Caused by CHCHD2 Loss-of-Function Mutation p.T61I. Neurobiol. Aging 2019, 75, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-T.; Huang, X.; Nguyen, D.; Shammas, M.K.; Wu, B.P.; Dombi, E.; Springer, D.A.; Poulton, J.; Sekine, S.; Narendra, D.P. Loss of CHCHD2 and CHCHD10 Activates OMA1 Peptidase to Disrupt Mitochondrial Cristae Phenocopying Patient Mutations. Hum. Mol. Genet. 2020, 29, 1547–1567. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Hu, J.; Che, Y.; Liu, Y.; Luo, Z.; Cheng, J.; Han, Q.; He, H.; Zhou, Q. CHCHD2 and CHCHD10 Regulate Mitochondrial Dynamics and Integrated Stress Response. Cell Death Dis. 2022, 13, 156. [Google Scholar] [CrossRef]
- Straub, I.R.; Weraarpachai, W.; Shoubridge, E.A. Multi-OMICS Study of a CHCHD10 Variant Causing ALS Demonstrates Metabolic Rewiring and Activation of Endoplasmic Reticulum and Mitochondrial Unfolded Protein Responses. Hum. Mol. Genet. 2021, 30, 687–705. [Google Scholar] [CrossRef]
- Liu, Y.; Clegg, H.V.; Leslie, P.L.; Di, J.; Tollini, L.A.; He, Y.; Kim, T.-H.; Jin, A.; Graves, L.M.; Zheng, J.; et al. CHCHD2 Inhibits Apoptosis by Interacting with Bcl-x L to Regulate Bax Activation. Cell Death Differ. 2015, 22, 1035–1046. [Google Scholar] [CrossRef]
- Aras, S.; Purandare, N.; Gladyck, S.; Somayajulu-Nitu, M.; Zhang, K.; Wallace, D.C.; Grossman, L.I. Mitochondrial Nuclear Retrograde Regulator 1 (MNRR1) Rescues the Cellular Phenotype of MELAS by Inducing Homeostatic Mechanisms. Proc. Natl. Acad. Sci. USA 2020, 117, 32056–32065. [Google Scholar] [CrossRef]
- Grossman, L.I.; Purandare, N.; Arshad, R.; Gladyck, S.; Somayajulu, M.; Hüttemann, M.; Aras, S. MNRR1, a Biorganellar Regulator of Mitochondria. Oxidative Med. Cell. Longev. 2017, 2017, 6739236. [Google Scholar] [CrossRef]
- Purandare, N.; Somayajulu, M.; Hüttemann, M.; Grossman, L.I.; Aras, S. The Cellular Stress Proteins CHCHD10 and MNRR1 (CHCHD2): Partners in Mitochondrial and Nuclear Function and Dysfunction. J. Biol. Chem. 2018, 293, 6517–6529. [Google Scholar] [CrossRef]
- Genin, E.C.; Bannwarth, S.; Lespinasse, F.; Ortega-Vila, B.; Fragaki, K.; Itoh, K.; Villa, E.; Lacas-Gervais, S.; Jokela, M.; Auranen, M.; et al. Loss of MICOS Complex Integrity and Mitochondrial Damage, but Not TDP-43 Mitochondrial Localisation, Are Likely Associated with Severity of CHCHD10-Related Diseases. Neurobiol. Dis. 2018, 119, 159–171. [Google Scholar] [CrossRef]
- Woo, J.-A.A.; Liu, T.; Trotter, C.; Fang, C.C.; De Narvaez, E.; LePochat, P.; Maslar, D.; Bukhari, A.; Zhao, X.; Deonarine, A.; et al. Loss of Function CHCHD10 Mutations in Cytoplasmic TDP-43 Accumulation and Synaptic Integrity. Nat. Commun. 2017, 8, 15558. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Woo, J.-A.A.; Bukhari, M.Z.; Wang, X.; Yan, Y.; Buosi, S.C.; Ermekbaeva, A.; Sista, A.; Kotsiviras, P.; LePochat, P.; et al. Modulation of Synaptic Plasticity, Motor Unit Physiology, and TDP-43 Pathology by CHCHD10. Acta Neuropathol. Commun. 2022, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “Dying-Back” Phenomenon of Motor Neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and Edaravone: A Tale of Two Amyotrophic Lateral Sclerosis Drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Siniscalchi, A.; Bonci, A.; Mercuri, N.B.; Bernardi, G. Effects of Riluzole on Rat Cortical Neurones: An in Vitro Electrophysiological Study: Riluzole in the Cerebral Cortex. Br. J. Pharmacol. 1997, 120, 225–230. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stevenson, A.; Yates, D.M.; Manser, C.; De Vos, K.J.; Vagnoni, A.; Leigh, P.N.; McLoughlin, D.M.; Miller, C.C.J. Riluzole Protects against Glutamate-Induced Slowing of Neurofilament Axonal Transport. Neurosci. Lett. 2009, 454, 161–164. [Google Scholar] [CrossRef]
- Koh, J.-Y.; Kim, D.-K.; Hwang, J.Y.; Kim, Y.H.; Seo, J.H. Antioxidative and Proapoptotic Effects of Riluzole on Cultured Cortical Neurons. J. Neurochem. 1999, 72, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.K. Riluzole but Not Melatonin Ameliorates Acute Motor Neuron Degeneration and Moderately Inhibits SOD1-Mediated Excitotoxicity Induced Disrupted Mitochondrial Ca2+ Signaling in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2017, 10, 295. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Xu, Z.-F.; Liu, W.; Xu, B.; Yang, H.-B.; Wei, Y.-G. Riluzole-Triggered GSH Synthesis via Activation of Glutamate Transporters to Antagonize Methylmercury-Induced Oxidative Stress in Rat Cerebral Cortex. Oxidative Med. Cell. Longev. 2012, 2012, 534705. [Google Scholar] [CrossRef]
- Abe, K.; Yuki, S.; Kogure, K. Strong Attenuation of Ischemic and Postischemic Brain Edema in Rats by a Novel Free Radical Scavenger. Stroke 1988, 19, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S. Neuroprotective Effects of Edaravone: Recent Insights. J. Neurol. Sci. 2013, 331, 177. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D. Edaravone: A New Drug Approved for ALS. Cell 2017, 171, 725. [Google Scholar] [CrossRef]
- Valko, K.; Ciesla, L. Amyotrophic Lateral Sclerosis. In Progress in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; Volume 58, pp. 63–117. [Google Scholar]
- Li, Q.; Qiu, Z.; Lu, Y.; Lu, P.; Wen, J.; Wang, K.; Zhao, X.; Li, R.; Zhang, H.; Zhang, Y.; et al. Edaravone Protects Primary-Cultured Rat Cortical Neurons from Ketamine-Induced Apoptosis via Reducing Oxidative Stress and Activating PI3K/Akt Signal Pathway. Mol. Cell. Neurosci. 2019, 100, 103399. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Tahara, M.; Todo, S. The Novel Antioxidant Edaravone: From Bench to Bedside. Cardiovasc. Ther. 2008, 26, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A.; Fang, T.; De Marchi, F.; Neel, D.; Van Weehaeghe, D.; Berry, J.D.; Paganoni, S. Pharmacotherapy for Amyotrophic Lateral Sclerosis: A Review of Approved and Upcoming Agents. Drugs 2022, 82, 1367–1388. [Google Scholar] [CrossRef]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Macklin, E.A.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; et al. Long-term Survival of Participants in the CENTAUR Trial of Sodium Phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve 2021, 63, 31–39. [Google Scholar] [CrossRef]
- Sun, Y.; Li, X.; Bedlack, R. An Evaluation of the Combination of Sodium Phenylbutyrate and Taurursodiol for the Treatment of Amyotrophic Lateral Sclerosis. Expert Rev. Neurother. 2023, 23, 1–7. [Google Scholar] [CrossRef]
- Fels, J.A.; Dash, J.; Leslie, K.; Manfredi, G.; Kawamata, H. Effects of PB-TURSO on the Transcriptional and Metabolic Landscape of Sporadic ALS Fibroblasts. Ann. Clin. Transl. Neurol. 2022, 9, 1551–1564. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate–Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Schumann, P.; Weydt, P.; Petri, S.; Koc, Y.; Spittel, S.; Bernsen, S.; Günther, R.; Weishaupt, J.H.; Dreger, M.; et al. Neurofilament Light-chain Response during Therapy with Antisense Oligonucleotide Tofersen in SOD1 -related ALS: Treatment Experience in Clinical Practice. Muscle Nerve 2023, 67, 515–521. [Google Scholar] [CrossRef] [PubMed]
- McCampbell, A.; Cole, T.; Wegener, A.J.; Tomassy, G.S.; Setnicka, A.; Farley, B.J.; Schoch, K.M.; Hoye, M.L.; Shabsovich, M.; Sun, L.; et al. Antisense Oligonucleotides Extend Survival and Reverse Decrement in Muscle Response in ALS Models. J. Clin. Investig. 2018, 128, 3558–3567. [Google Scholar] [CrossRef]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, P.; Thompson, J.L.P.; Levy, G.; Buchsbaum, R.; Shefner, J.; Krivickas, L.S.; Katz, J.; Rollins, Y.; Barohn, R.J.; Jackson, C.E.; et al. Phase II Trial of CoQ10 for ALS Finds Insufficient Evidence to Justify Phase III. Ann. Neurol. 2009, 66, 235–244. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Name | Affected Mitochondrial Functions | ||
|---|---|---|---|---|
| Mitochondrial dysfunctions in ALS | ALS1 | SOD1 | Cu/Zn superoxide dismutase 1 | Aggregates, increased oxidative stress, impaired RC activity, disrupted mitochonrial dynamics and morpholgy, disrupted mitophagy, disrupted ER-mitochondria contacts and calcium homestasis |
| ALS-FTD1 | C9ORF72 | Chromosome 9 open reading frame 72 | Increased oxygen consumption, mitochondrial hyperpolarization, impaired RC activity, disrupted mitophagy, disrupted mitochonrial dynamics and morphology, disrupted ER-mitochondria contacts | |
| ALS10 | TARDBP/TDP-43 | Trans-activating response region DNA-binding protein 43 | Agreggates, accumulation in mitochondria, increased oxidative stress, disrupted mitochondrial dynamics and morphology, disrupted mitophagy, disrupted ER-mitochondria contacts and calcium homestasis | |
| ALS6 | FUS | Fused in Sarcoma | Agreggates, increased oxydative stress, impaired ATP production, disrupted mitophagy, disrupted ER-mitochondria contacts and calcium homeostasis | |
| ALS2 | ALS2 | Alsin | Disrupted mitophagy, disrupted mitochondrial morphology, disrupted endosomal and mitochondrial transport | |
| ALS8 | VAPB | Vesicle-associated membrane protein-associated protein B | Disrupted ER-mitochondria contacts and calcium homeostasis, disrupted anterograde axonal transport | |
| ALS12 | OPTN | Optineurin | Inclusions, disrupted mitophagy | |
| ALS16 | SIGMAR1 | Sigma-1 receptor | Agreggates, disrupted mitochondrial dynamic, disrupted ER-mitochondria contacts and calcium homeostasiss, disrupted axonal transport | |
| ALS-FTD3 | SQSTM1 | p62/Sequestosome 1 | Agreggates, increased oxidative stress, decreased mitochondrial respiration, disrupted mitophagy, disrupted mitochondrial membrane potential | |
| ALS14 | VCP | Valosin-containing protein | Disrupted ER-mitochondria contacts and calcium homeostasis, disrupted mitophagy | |
| ALS-FTD2 | CHCHD10 | Coiled-coil-helix-coiled-coil-helix domain containing 10 | Aggregates, disrupted mitochondrial dynamics and morphology, decreased mitochondrial respiration, disrupted mitochondrial potential, mt-ISR activation |
| Name | FDA Approved (Year) | Mechanisms |
|---|---|---|
| Riluzole | 1995 | Glutamate antagonist that inhibits glutamate release and protein kinase c, regulates intracellular events that follow transmitter binding to excitatory receptors and regulates calcium flux |
| Edaravone | 2017 | Antioxidant that removes oxygen radicals and eliminates lipid peroxides in the central nervous system |
| Sodium phenylbutyrate and Taurursodiol (PB/TURSO) | 2022 | Decreases ER stress and mitochondrial dysfunction |
| Tofersen | 2023 | Antisense oligonucleotide that degrades SOD1 messenger RNA and reduces SOD1 protein levels |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genin, E.C.; Abou-Ali, M.; Paquis-Flucklinger, V. Mitochondria, a Key Target in Amyotrophic Lateral Sclerosis Pathogenesis. Genes 2023, 14, 1981. https://doi.org/10.3390/genes14111981
Genin EC, Abou-Ali M, Paquis-Flucklinger V. Mitochondria, a Key Target in Amyotrophic Lateral Sclerosis Pathogenesis. Genes. 2023; 14(11):1981. https://doi.org/10.3390/genes14111981
Chicago/Turabian StyleGenin, Emmanuelle C., Mélanie Abou-Ali, and Véronique Paquis-Flucklinger. 2023. "Mitochondria, a Key Target in Amyotrophic Lateral Sclerosis Pathogenesis" Genes 14, no. 11: 1981. https://doi.org/10.3390/genes14111981
APA StyleGenin, E. C., Abou-Ali, M., & Paquis-Flucklinger, V. (2023). Mitochondria, a Key Target in Amyotrophic Lateral Sclerosis Pathogenesis. Genes, 14(11), 1981. https://doi.org/10.3390/genes14111981