
Impacts of Population Size and Domestication Process on Genetic Diversity and Genetic Load in Genus Ovis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collecting
2.2. SNP Calling
2.3. Estimating Index of Genetic Diversity and Calculating Runs of Homozygosity (ROH)
2.4. Estimates of Effective Population Size (Ne)
2.5. Estimate of Genetic Load
3. Results
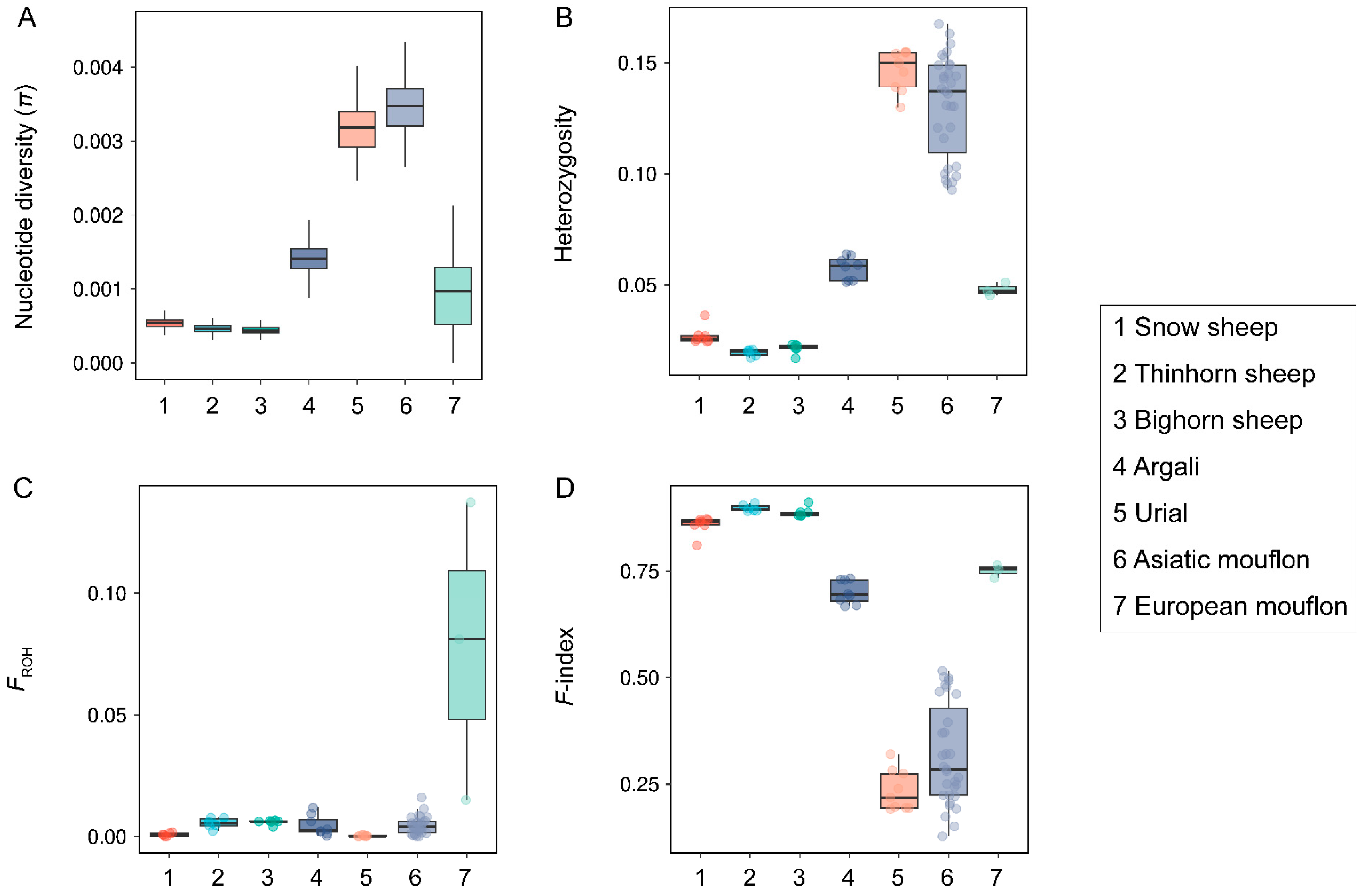
3.1. Genetic Diversity Pattern
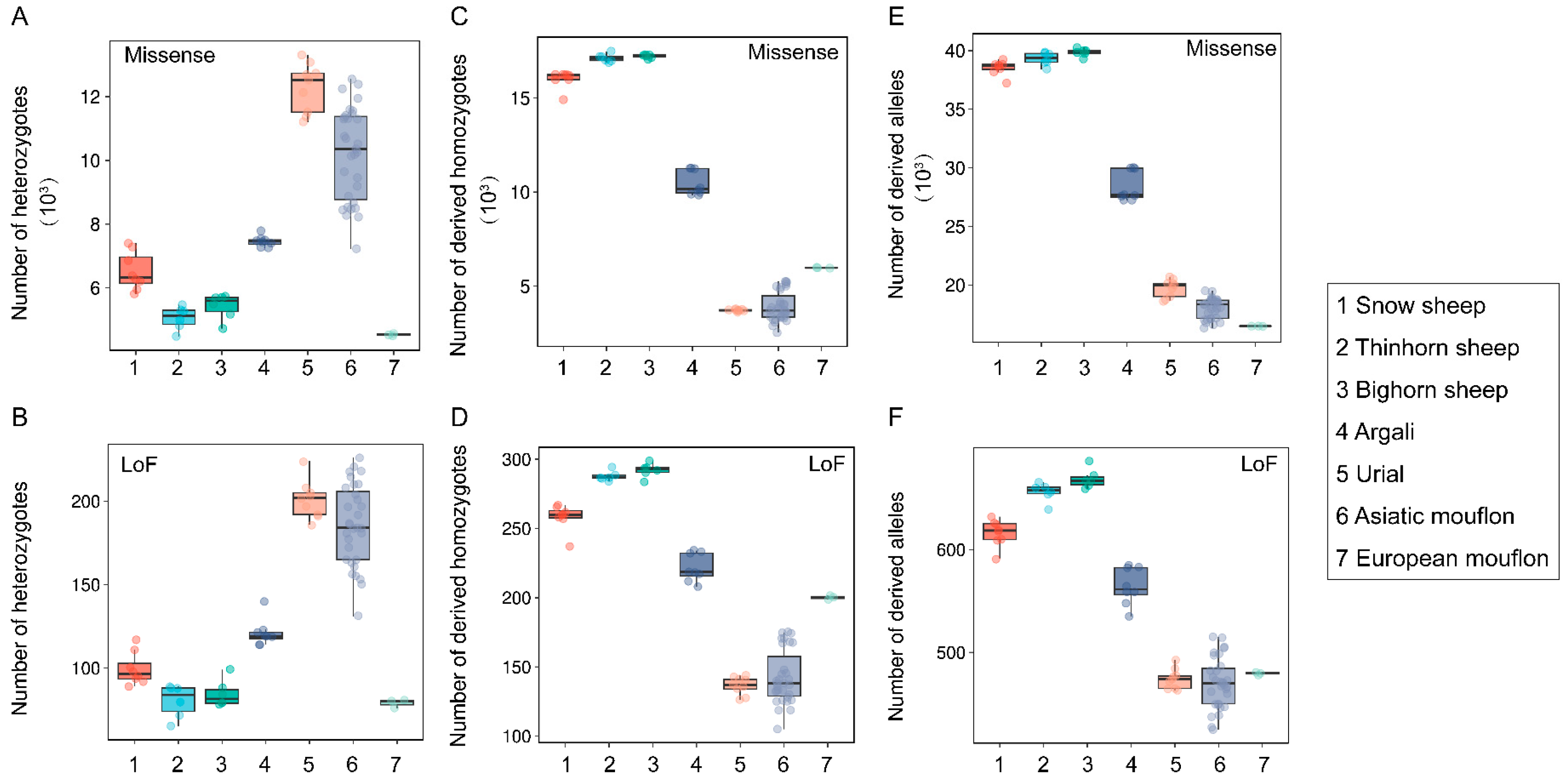
3.2. Genetic Load Comparisons among Wild Species within the Genus Ovis
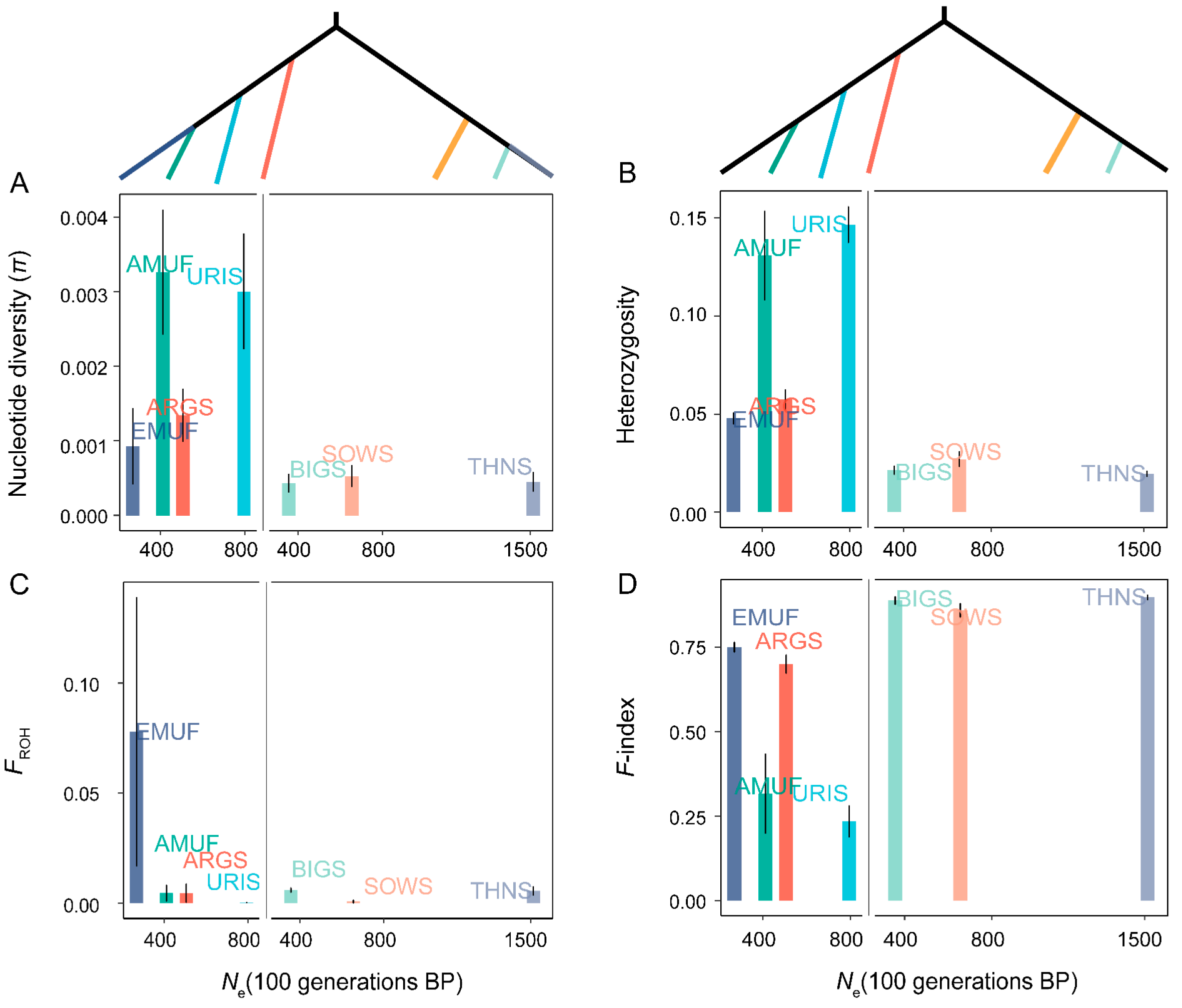
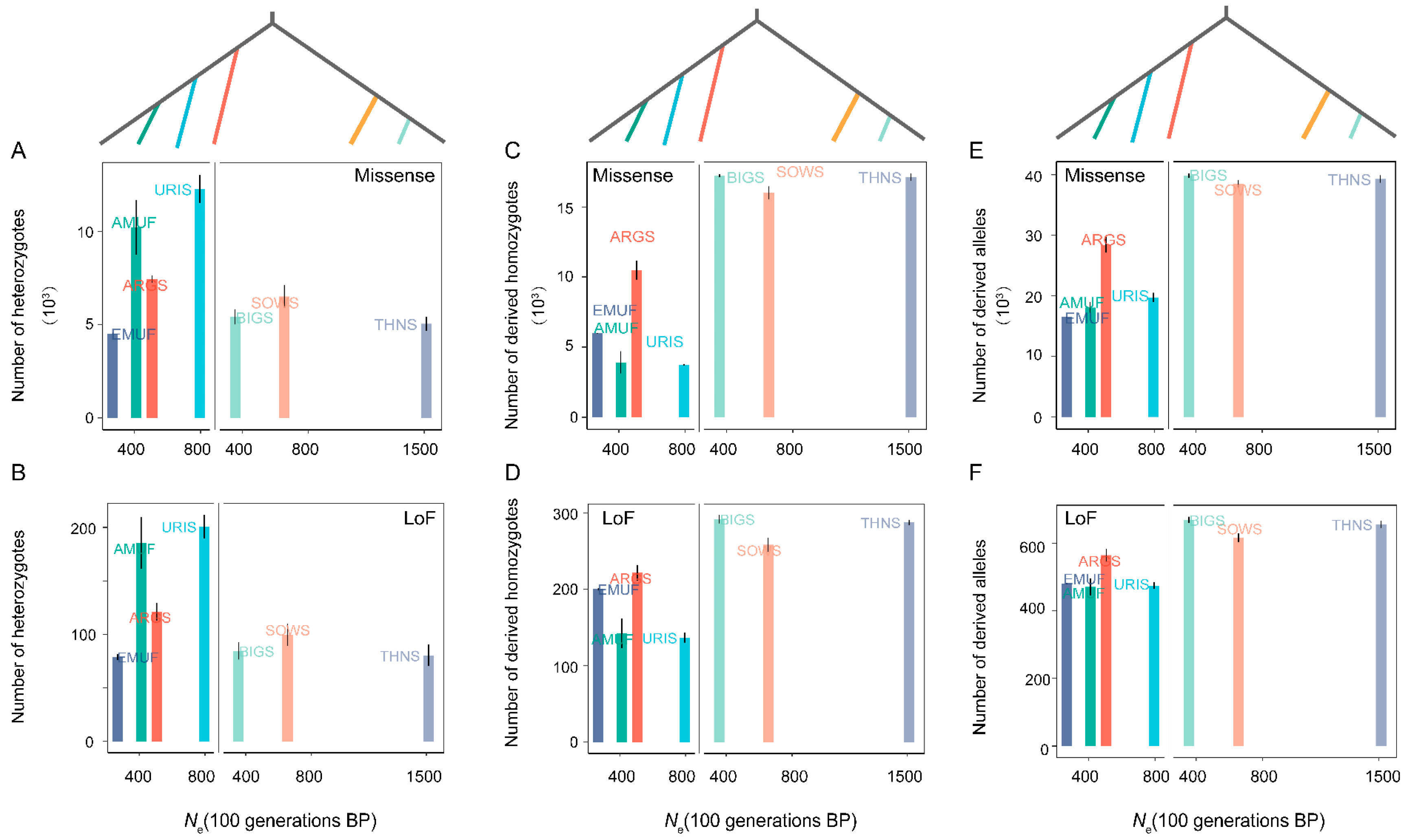
3.3. Genetic Load and Effective Population Size (Ne)
3.4. Relationships of Domestication and Improvement Process with Genetic Diversity and Genetic Load
4. Discussion
4.1. Genetic Diversity and Mutation Burden among Wild Species
4.2. Ne and Genetic Load
4.3. Cost of Domestication
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Code Availability:
References
- Leroy, T.; Rousselle, M.; Tilak, M.-K.; Caizergues, A.E.; Scornavacca, C.; Recuerda, M.; Fuchs, J.; Illera, J.C.; De Swardt, D.H.; Blanco, G.; et al. Island songbirds as windows into evolution in small populations. Curr. Biol. 2021, 31, 1303–1310.e4. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.E.; Woodworth, L.M.; Nurthen, R.K.; Gilligan, D.M.; Briscoe, D.A.; Frankham, R. Relationships between population size and loss of genetic diversity: Comparisons of experimental results with theoretical predictions. Conserv. Genet. 2000, 1, 33–43. [Google Scholar] [CrossRef]
- Buffalo, V. Quantifying the relationship between genetic diversity and population size suggests natural selection cannot explain Lewontin’s Paradox. eLife 2021, 10, e67509. [Google Scholar] [CrossRef] [PubMed]
- Dussex, N.; van der Valk, T.; Morales, H.E.; Wheat, C.W.; Díez-del-Molino, D.; von Seth, J.; Foster, Y.; Kutschera, V.E.; Guschanski, K.; Rhie, A.; et al. Population genomics of the critically endangered kākāpō. Cell Genom. 2021, 1, 100002. [Google Scholar] [CrossRef]
- Robinson, J.A.; Brown, C.; Kim, B.Y.; Lohmueller, K.E.; Wayne, R.K. Purging of Strongly Deleterious Mutations Explains Long-Term Persistence and Absence of Inbreeding Depression in Island Foxes. Curr. Biol. 2018, 28, 3487–3494. [Google Scholar] [CrossRef] [PubMed]
- Moyers, B.T.; Morrell, P.L.; McKay, J.K. Genetic Costs of Domestication and Improvement. J. Hered. 2017, 109, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-S.; Zhang, J.-J.; Guo, X.; Li, M.; Meyer, R.; Ashari, H.; Zheng, Z.-Q.; Wang, S.; Peng, M.-S.; Jiang, Y.; et al. Large-scale genomic analysis reveals the genetic cost of chicken domestication. BMC Biol. 2021, 19, 118. [Google Scholar] [CrossRef]
- Marsden, C.D.; Ortega-Del Vecchyo, D.; O’Brien, D.P.; Taylor, J.F.; Ramirez, O.; Vilà, C.; Marques-Bonet, T.; Schnabel, R.D.; Wayne, R.K.; Lohmueller, K.E. Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc. Natl. Acad. Sci. USA 2016, 113, 152–157. [Google Scholar] [CrossRef]
- Gaut, B.S.; Seymour, D.K.; Liu, Q.; Zhou, Y. Demography and its effects on genomic variation in crop domestication. Nat. Plants 2018, 4, 512–520. [Google Scholar] [CrossRef]
- Samayoa, L.F.; Olukolu, B.A.; Yang, C.J.; Chen, Q.; Stetter, M.G.; York, A.M.; Sanchez-Gonzalez, J.d.J.; Glaubitz, J.C.; Bradbury, P.J.; Romay, M.C.; et al. Domestication reshaped the genetic basis of inbreeding depression in a maize landrace compared to its wild relative, teosinte. PLoS Genet. 2021, 17, e1009797. [Google Scholar] [CrossRef]
- Chen, Z.-H.; Xu, Y.-X.; Xie, X.-L.; Wang, D.-F.; Aguilar-Gómez, D.; Liu, G.-J.; Li, X.; Esmailizadeh, A.; Rezaei, V.; Kantanen, J.; et al. Whole-genome sequence analysis unveils different origins of European and Asiatic mouflon and domestication-related genes in sheep. Commun. Biol. 2021, 4, 1307. [Google Scholar] [CrossRef] [PubMed]
- Barbato, M.; Hailer, F.; Orozco-terWengel, P.; Kijas, J.; Mereu, P.; Cabras, P.; Mazza, R.; Pirastru, M.; Bruford, M.W. Genomic signatures of adaptive introgression from European mouflon into domestic sheep. Sci. Rep. 2017, 7, 7623. [Google Scholar] [CrossRef] [PubMed]
- Hermans, W.A. The European mouflon. Ovis musimon. Tijdschr. Voor Diergeneeskd. 1996, 121, 515–517. [Google Scholar]
- Chessa, B.; Pereira, F.; Arnaud, F.; Amorim, A.; Goyache, F.; Mainland, I.; Kao, R.R.; Pemberton, J.M.; Beraldi, D.; Stear, M.J.; et al. Revealing the History of Sheep Domestication Using Retrovirus Integrations. Science 2009, 324, 532–536. [Google Scholar] [CrossRef]
- Maher, L.A.; Richter, T.; Stock, J.T. The Pre-Natufian Epipaleolithic: Long-term Behavioral Trends in the Levant. Evol. Anthropol. Issues News Rev. 2012, 21, 69–81. [Google Scholar] [CrossRef]
- Zeder, M.A. Domestication and early agriculture in the Mediterranean Basin: Origins, diffusion, and impact. Proc. Natl. Acad. Sci. USA 2008, 105, 11597–11604. [Google Scholar] [CrossRef]
- Li, X.; Yang, J.; Shen, M.; Xie, X.-L.; Liu, G.-J.; Xu, Y.-X.; Lv, F.-H.; Yang, H.; Yang, Y.-L.; Liu, C.-B.; et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 2020, 11, 2815. [Google Scholar] [CrossRef]
- Kalds, P.; Zhou, S.; Gao, Y.; Cai, B.; Huang, S.; Chen, Y.; Wang, X. Genetics of the phenotypic evolution in sheep: A molecular look at diversity-driving genes. Genet. Sel. Evol. 2022, 54, 61. [Google Scholar] [CrossRef]
- Lv, F.-H.; Cao, Y.-H.; Liu, G.-J.; Luo, L.-Y.; Lu, R.; Liu, M.-J.; Li, W.-R.; Zhou, P.; Wang, X.-H.; Shen, M.; et al. Whole-genome resequencing of worldwide wild and domestic sheep elucidates genetic diversity, introgression and agronomically important loci. Mol. Biol. Evol. 2022, 39, msab353. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes; CRAN (The Comprehensive R Archive Network): Vienna, Austria, 2018. [Google Scholar]
- Gorssen, W.; Meyermans, R.; Janssens, S.; Buys, N. A publicly available repository of ROH islands reveals signatures of selection in different livestock and pet species. Genet. Sel. Evol. 2021, 53, 2. [Google Scholar] [CrossRef]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. How to study runs of homozygosity using PLINK? A guide for analyzing medium density SNP data in livestock and pet species. BMC Genom. 2020, 21, 94. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wang, L.; Liu, J.; Deng, T.; Yan, H.; Zhang, L.; Liu, X.; Gao, H.; Hou, X.; Wang, L.; et al. Estimation of inbreeding and identification of regions under heavy selection based on runs of homozygosity in a Large White pig population. J. Anim. Sci. Biotechnol. 2020, 11, 46. [Google Scholar] [CrossRef]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef]
- Sved, J.A. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 1971, 2, 125–141. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Humble, E.; Stoffel, M.A.; Dicks, K.; Ball, A.D.; Gooley, R.M.; Chuven, J.; Pusey, R.; Remeithi, M.A.; Koepfli, K.-P.; Pukazhenthi, B.; et al. Conservation management strategy impacts inbreeding and mutation load in scimitar-horned oryx. Proc. Natl. Acad. Sci. USA 2023, 120, e2210756120. [Google Scholar] [CrossRef]
- Mathur, S.; DeWoody, J.A. Genetic load has potential in large populations but is realized in small inbred populations. Evol. Appl. 2021, 14, 1540–1557. [Google Scholar] [CrossRef] [PubMed]
- Bertorelle, G.; Raffini, F.; Bosse, M.; Bortoluzzi, C.; Iannucci, A.; Trucchi, E.; Morales, H.E.; van Oosterhout, C. Genetic load: Genomic estimates and applications in non-model animals. Nat. Rev. Genet. 2022, 23, 492–503. [Google Scholar] [CrossRef]
- Grossen, C.; Guillaume, F.; Keller, L.F.; Croll, D. Purging of highly deleterious mutations through severe bottlenecks in Alpine ibex. Nat. Commun. 2020, 11, 1001. [Google Scholar] [CrossRef]
- Morrell, P.L.; Buckler, E.S.; Ross-Ibarra, J. Crop genomics: Advances and applications. Nat. Rev. Genet. 2012, 13, 85–96. [Google Scholar] [CrossRef]
- Alberto, F.J.; Boyer, F.; Orozco-terWengel, P.; Streeter, I.; Servin, B.; de Villemereuil, P.; Benjelloun, B.; Librado, P.; Biscarini, F.; Colli, L.; et al. Convergent genomic signatures of domestication in sheep and goats. Nat. Commun. 2018, 9, 813. [Google Scholar] [CrossRef]
- Cheng, H.; Zhang, Z.; Wen, J.; Lenstra, J.A.; Heller, R.; Cai, Y.; Guo, Y.; Li, M.; Li, R.; Li, W.; et al. Long divergent haplotypes introgressed from wild sheep are associated with distinct morphological and adaptive characteristics in domestic sheep. PLoS Genet. 2023, 19, e1010615. [Google Scholar] [CrossRef]
- IUCN. The IUCN Red List of Threatened Species. 2020. Available online: https://www.iucnredlist.org (accessed on 25 June 2023).
- Orellana, M.-R.; López-Pujol, J.; Blanché, C.; Bosch, M. Relationships between heterozygosity and fitness in the Iberian threatened larkspur Delphinium bolosii (Ranunculaceae). Flora Morphol. Distrib. Funct. Ecol. 2007, 202, 161–168. [Google Scholar] [CrossRef]
- Grueber, C.E.; Wallis, G.P.; Jamieson, I.G. Heterozygosity–fitness correlations and their relevance to studies on inbreeding depression in threatened species. Mol. Ecol. 2008, 17, 3978–3984. [Google Scholar] [CrossRef]
- Upadhyay, M.; Kunz, E.; Sandoval-Castellanos, E.; Hauser, A.; Krebs, S.; Graf, A.; Blum, H.; Dotsev, A.; Okhlopkov, I.; Shakhin, A.; et al. Whole genome sequencing reveals a complex introgression history and the basis of adaptation to subarctic climate in wild sheep. Mol. Ecol. 2021, 30, 6701–6717. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, H.R.; Naderi, S.; Chintauan-Marquier, I.C.; Taberlet, P.; Virk, A.T.; Naghash, H.R.; Rioux, D.; Kaboli, M.; Pompanon, F. Evolution and taxonomy of the wild species of the genus Ovis (Mammalia, Artiodactyla, Bovidae). Mol. Phylogenet. Evol. 2010, 54, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.-H.; Peng, W.-F.; Yang, J.; Zhao, Y.-X.; Li, W.-R.; Liu, M.-J.; Ma, Y.-H.; Zhao, Q.-J.; Yang, G.-L.; Wang, F.; et al. Mitogenomic Meta-Analysis Identifies Two Phases of Migration in the History of Eastern Eurasian Sheep. Mol. Biol. Evol. 2015, 32, 2515–2533. [Google Scholar] [CrossRef] [PubMed]
- Brivio, F.; Ciuti, S.; Pipia, A.; Grignolio, S.; Apollonio, M. Livestock displace European mouflon from optimal foraging sites. Eur. J. Wildl. Res. 2022, 68, 30. [Google Scholar] [CrossRef]
- Ptak, G.; Clinton, M.; Barboni, B.; Muzzeddu, M.; Cappai, P.; Tischner, M.; Loi, P. Preservation of the Wild European Mouflon: The First Example of Genetic Management Using a Complete Program of Reproductive Biotechnologies. Biol. Reprod. 2002, 66, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Santiago, E.; Caballero, A. Prediction and estimation of effective population size. Heredity 2016, 117, 193–206. [Google Scholar] [CrossRef]
- Kardos, M.; Zhang, Y.; Parsons, K.M.; Kang, H.; Xu, X.; Liu, X.; Matkin, C.O.; Zhang, P.; Ward, E.J.; Hanson, M.B.; et al. Inbreeding depression explains killer whale population dynamics. Nat. Ecol. Evol. 2023, 7, 675–686. [Google Scholar] [CrossRef]
- Lu, J.; Tang, T.; Tang, H.; Huang, J.; Shi, S.; Wu, C.-I. The accumulation of deleterious mutations in rice genomes: A hypothesis on the cost of domestication. Trends Genet. 2006, 22, 126–131. [Google Scholar] [CrossRef]
- Ciani, E.; Lasagna, E.; D’Andrea, M.; Alloggio, I.; Marroni, F.; Ceccobelli, S.; Delgado Bermejo, J.; Sarti, F.; Kijas, J.; Lenstra, J.; et al. Merino and Merino-derived sheep breeds: A genome-wide intercontinental study. Genet. Sel. Evol. 2015, 47, 64. [Google Scholar] [CrossRef]
- Dussex, N.; Morales, H.E.; Grossen, C.; Dalén, L.; van Oosterhout, C. Purging and accumulation of genetic load in conservation. Trends Ecol. Evol. 2023, 38, 961–969. [Google Scholar] [CrossRef] [PubMed]
- van der Valk, T.; Gonda, C.M.; Silegowa, H.; Almanza, S.; Sifuentes-Romero, I.; Hart, T.; Hart, J.; Detwiler, K.M.; Guschanski, K. The genome of the endangered Dryas monkey provides new insights into the evolutionary history of the vervets. Mol. Biol. Evol. 2019, 37, 183–194. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed/Species | Abbr. | Group | Number of Individual | Country | Data |
|---|---|---|---|---|---|
| Europe Mouflon (Ovis musimon) | EMUF | Wild species | 3 | Finland | WGS |
| Asiatic Mouflon (Ovis orientalis) | AMUF | Wild species | 31 | Iran | WGS |
| Urial (Ovis vignei) | URIS | Wild species | 9 | Iran | WGS |
| Argali (Ovis ammon) | ARGS | Wild species | 8 | China | WGS |
| Snow (Ovis nivicola) | SOWS | Wild species | 8 | Russia | WGS |
| Thinhorn (Ovis dalli) | THNS | Wild species | 6 | Canada | WGS |
| Bighorn (Ovis canadensis) | BIGS | Wild species | 6 | Canada | WGS |
| Tibetan | TBT | Native breed | 15 | China | WGS |
| Finn | FIN | Native breed | 10 | Finland | WGS |
| Shetland | SHE | Native breed | 7 | The Netherlands | WGS |
| Chinese Merino (super fine wool) | MSF | Improved breed | 10 | China | WGS |
| Chinese Merino (fine wool) | MFW | Improved breed | 10 | China | WGS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Salehian-Dehkordi, H.; Suo, L.; Lv, F. Impacts of Population Size and Domestication Process on Genetic Diversity and Genetic Load in Genus Ovis. Genes 2023, 14, 1977. https://doi.org/10.3390/genes14101977
Wang D, Salehian-Dehkordi H, Suo L, Lv F. Impacts of Population Size and Domestication Process on Genetic Diversity and Genetic Load in Genus Ovis. Genes. 2023; 14(10):1977. https://doi.org/10.3390/genes14101977
Chicago/Turabian StyleWang, Dongfeng, Hosein Salehian-Dehkordi, Langda Suo, and Fenghua Lv. 2023. "Impacts of Population Size and Domestication Process on Genetic Diversity and Genetic Load in Genus Ovis" Genes 14, no. 10: 1977. https://doi.org/10.3390/genes14101977
APA StyleWang, D., Salehian-Dehkordi, H., Suo, L., & Lv, F. (2023). Impacts of Population Size and Domestication Process on Genetic Diversity and Genetic Load in Genus Ovis. Genes, 14(10), 1977. https://doi.org/10.3390/genes14101977