
Predictive Clinical and Biological Criteria for Gene Panel Positivity in Suspected Inherited Autoinflammatory Diseases: Insights from a Case–Control Study
 ,
,
Abstract
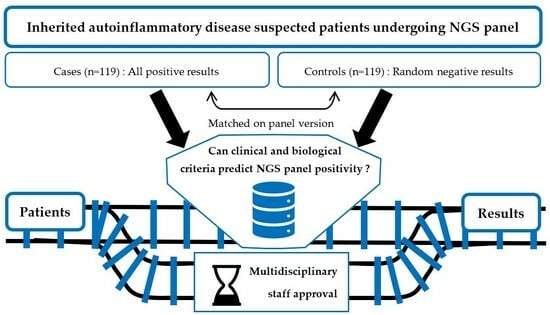
:
1. Introduction
2. Methods
2.1. Selection of Cases and Controls
2.2. Clinical and Biological Characteristics
2.3. Study Design
2.4. Statistical Analysis
2.5. Patients
3. Results
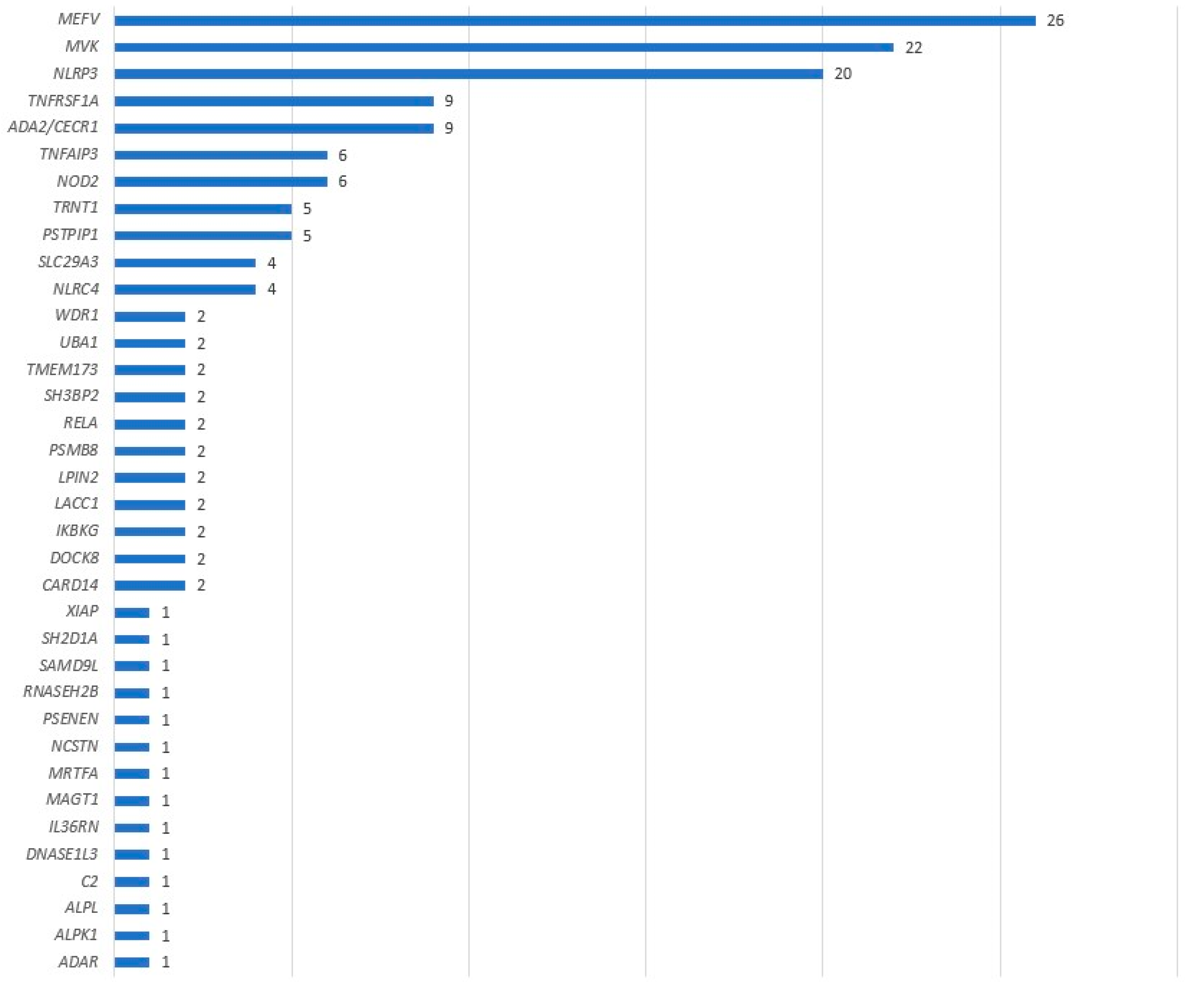
3.1. Positive Panel Results
3.2. Clinical and Biological Characteristics
3.3. Diagnostic and Predictive Values
3.4. Panel Subgroup Analyses
3.5. Exploratory Analyses
4. Discussion
4.1. Clinical and Biological Criteria as Positive Panel Predictors?
4.2. Limitations
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviations | Meaning |
| CI | Confidence Interval |
| CRP | C-Reactive Protein |
| FMF | Familial Mediterranean Fever |
| HLH | Hemophagocytic LymphoHistiocytosis |
| HS | Hidradenitis Suppurativa |
| IBD | Inflammatory Bowl Diseases |
| ISSAIDS | International Society of Systemic Auto-Inflammatory Diseases |
| MDS | Myelo-Dysplasic Syndrome |
| NGS | Next-Generation Sequencing |
| NPV | Negative Preditive Value |
| OR | Odds Ratio |
| PPV | Positive Predictive Value |
| SAA | Serum Amyloid A |
| Se | Sensibility |
| Sp | Specificity |
| VUS | Variant of Unknown Significance |
| WES | Whole Exome Sequencing |
| WGS | Whole Genome Sequencing |
References
- Krainer, J.; Siebenhandl, S. Systemic autoinflammatory diseases. J. Autoimmun. 2020, 109, 102421. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.B.; Shohat, M. Familial Mediterranean fever in Armenians: Autosomal recessive inheritance with high gene frequency. Am. J. Med. Genet. 1989, 34, 168–172. [Google Scholar] [CrossRef] [PubMed]
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat. Genet. 1997, 17, 25–31. [Google Scholar] [CrossRef] [PubMed]
- The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997, 90, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Chitnis, N. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Boursier, G.; Rittore, C. Positive Impact of Expert Reference Center Validation on Performance of Next-Generation Sequencing for Genetic Diagnosis of Autoinflammatory Diseases. J. Clin. Med. 2019, 8, 1729. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Meckfessel, M.H. Autoinflammatory disorders, pain, and neural regulation of inflammation. Dermatol. Clin. 2013, 31, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Gattorno, M. Neurological manifestations in autoinflammatory diseases. Clin. Exp. Rheumatol. 2018, 36 (Suppl. S110), 61–67. [Google Scholar] [PubMed]
- Georgin-Lavialle, S.; Terrier, B. Further characterization of clinical and laboratory features in VEXAS syndrome: Large-scale analysis of a multicentre case series of 116 French patients. Br. J. Dermatol. 2022, 186, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Similuk, M.N.; Yan, J. Clinical exome sequencing of 1000 families with complex immune phenotypes: Toward comprehensive genomic evaluations. J. Allergy Clin. Immunol. 2022, 150, 947–954. [Google Scholar] [CrossRef] [PubMed]
- McCreary, D.; Omoyinmi, E. A rapid turnaround gene panel for severe autoinflammation: Genetic results within 48 hours. Front. Immunol. 2022, 13, 998967. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Kawashima, Y. NLRP3 mutation and cochlear autoinflammation cause syndromic and nonsyndromic hearing loss DFNA34 responsive to anakinra therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E7766–E7775. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, C.; Omoyinmi, E. Monogenic mimics of Behçet’s disease in the young. Rheumatology 2019, 58, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Betrains, A.; Staels, F. Systemic autoinflammatory disease in adults. Autoimmun. Rev. 2021, 20, 102774. [Google Scholar] [CrossRef] [PubMed]
- Shinar, Y.; Ceccherini, I. ISSAID/EMQN Best Practice Guidelines for the Genetic Diagnosis of Monogenic Autoinflammatory Diseases in the Next-Generation Sequencing Era. Clin. Chem. 2020, 66, 525–536. [Google Scholar] [CrossRef] [PubMed]
- BiostaTGV. Available online: https://biostatgv.sentiweb.fr/ (accessed on 17 July 2023).
- MedCalc. Available online: www.medcalc.org (accessed on 17 July 2023).

{kind=link}
{kind=link}
| Characteristics | Cases n (%) | Controls n (%) | p | Characteristics | Cases n (%) | Controls n (%) | p |
|---|---|---|---|---|---|---|---|
| Age of onset | Digestive | ||||||
| mean (months), 95% CI | 96 (61; 131) | 137 (106; 168) | 0.09 | abscess | 2 (1.7) | 4 (3.4) | 0.68 |
| neonatal (<30 days old) § | 12/104 (11.5) | 1/107 (0.9) | 0.001 | hepatic cytolysis | 11 (9.2) | 14 (11.8) | 0.53 |
| early childhood (≤3 years old) § | 56/104 (53.8) | 30/107 (28.0) | 0.0001 | IBD | 2 (1.7) | 6 (5.0) | 0.28 |
| childhood (<18 years old) § | 94/104 (90.4) | 87/107 (81.3) | 0.06 | diarrhea/vomiting | 34 (28.6) | 34 (28.6) | 1 |
| adult (≥18 years old) § | 10/104 (9.6) | 20/107 (18.7) | 0.06 | abdominal pain | 47 (39.5) | 52 (43.7) | 0.51 |
| Inflammation | hemorrhage | 3 (2.5) | 7 (5.9) | 0.20 | |||
| fever | 86 (72.3) | 87 (73.1) | 0.88 | Cutaneous | |||
| crisis CRP elevation | 102 (85.7) | 98 (82.4) | 0.48 | aphthous ulceration * | 42 (35.3) | 42 (35.3) | 1 |
| Thoracic | erythema nodosum | 4 (3.4) | 7 (5.9) | 0.34 | |||
| chest pain | 25 (21.0) | 25 (21.0) | 1 | folliculitis, acne, HS | 18 (15.1) | 19 (16.0) | 0.86 |
| pleuro-pericarditis | 9 (7.6) | 9 (7.6) | 1 | lipodystrophia | 4 (3.4) | 4 (3.4) | 1 |
| pneumonia | 8 (6.7) | 8 (6.7) | 1 | livedo | 6 (5.0) | 6 (5.0) | 1 |
| Neurologic and sensorineural | maculopapular rash/urticaria | 42 (35.3) | 33 (27.7) | 0.21 | |||
| stroke | 3 (2.5) | 7 (5.9) | 0.20 | necrosis | 4 (3.4) | 6 (5.0) | 0.52 |
| cerebral calcifications | 2 (1.7) | 1 (0.8) | 1 | edema | 8 (6.7) | 9 (7.6) | 0.80 |
| headaches | 26 (21.8) | 36 (30.3) | 0.14 | pseudoerysipelas | 9 (7.6) | 7 (5.9) | 0.60 |
| conjunctivitis | 21 (17.6) | 13 (10.9) | 0.14 | psoriasis | 6 (5.0) | 4 (3.4) | 0.52 |
| encephalitis | 1 (0.8) | 2 (1.7) | 1 | pyoderma | 0 (0.0) | 3 (2.5) | 0.25 |
| epilepsy | 1 (0.8) | 3 (2.5) | 0.62 | tenosynovitis | 6 (5.0) | 6 (5.0) | 1 |
| meningitis | 8 (6.7) | 3 (2.5) | 0.12 | vasculitis/Raynaud | 14 (11.8) | 10 (8.4) | 0.39 |
| papillitis | 5 (4.2) | 0 (0.0) | 0.06 | Diverse | |||
| uveitis | 11 (9.2) | 9 (7.6) | 0.64 | failure to thrive | 24 (20.2) | 12 (10.1) | 0.03 |
| deafness | 19 (16.0) | 2 (1.7) | 0.0001 | adenopathy | 26 (21.8) | 30 (25.2) | 0.54 |
| intellectual disabilities | 6 (5.0) | 4 (3.4) | 0.52 | immunodeficiency | 6 (5.0) | 2 (1.7) | 0.28 |
| Locomotor | hepatomegaly | 3 (2.5) | 4 (3.4) | 1 | |||
| arthralgia | 73 (61.3) | 80 (67.2) | 0.34 | splenomegaly | 9 (7.6) | 5 (4.2) | 0.27 |
| arthritis | 42 (35.3) | 29 (24.4) | 0.07 | recurrent infections | 10 (8.4) | 14 (11.8) | 0.39 |
| myalgia/myositis | 32 (26.9) | 35 (29.4) | 0.67 | pharyngitis | 14 (11.8) | 26 (21.8) | 0.04 |
| distorting arthropathy | 5 (4.2) | 3 (2.5) | 0.72 | polychondritis | 2 (1.7) | 0 (0.0) | 0.50 |
| osteitis | 3 (2.5) | 6 (5.0) | 0.50 | HLH | 1 (0.8) | 6 (5.0) | 0.12 |
| Renal | MDS | 2 (1.7) | 1 (0.8) | 1 | |||
| amyloidosis | 6 (5.0) | 2 (1.7) | 0.28 | obesity | 4 (3.4) | 4 (3.4) | 1 |
| kidney failure | 10 (8.4) | 4 (3.4) | 0.10 | ||||
| proteinuria | 11 (9.2) | 5 (4.2) | 0.12 | ||||
| Combinations | Cases n (%) | Controls n (%) | p |
|---|---|---|---|
| NO fever and/or NO CRP elevation | 39 (32.8) | 38 (31.9) | 0.89 |
| NO fever and NO CRP elevation | 11 (9.2) | 15 (12.6) | 0.41 |
| Bipolar aphthosis | 8 (6.7) | 17 (14.3) | 0.06 |
| Hepatosplenomegaly | 14 (11.8) | 5 (4.2) | 0.03 |
| No other system than inflammatory | 1 (0.8) | 2 (1.7) | 1 |
| At least 1 system (except inflammatory) | 118 (99.2) | 117 (98.3) | 1 |
| At least 2 systems (except inflammatory) | 108 (90.9) | 109 (91.6) | 0.82 |
| At least 3 systems (except inflammatory) | 86 (72.3) | 93 (78.2) | 0.29 |
| At least 4 systems (except inflammatory) | 56 (47.1) | 55 (46.2) | 0.90 |
| At least 5 systems (except inflammatory) | 29 (24.4) | 30 (25.2) | 0.88 |
| At least 6 systems (except inflammatory) | 9 (7.6) | 8 (6.7) | 0.80 |
| All 7 systems (except inflammatory) | 2 (1.7) | 0 (0.0) | 0.50 |
| Mean of the number of systems involved (except inflammatory), 95% CI | 3.43 (0.51; 6.35) | 3.47 (0.74; 6.20) | 0.82 |
| Characteristics | Cases n (%) | Controls n (%) | OR (95% CI) | Se (%) (95% CI) | Sp (%) (95% CI) | PPV (%) * (95% CI) | NPV (%) * (95% CI) | Accuracy (%) * (95% CI) |
|---|---|---|---|---|---|---|---|---|
| Neonatal onset § | 12/104 (11.5) | 1/107 (0.9) | 13.83 (1.76; 108.38) | 11.5 (6.1; 19.3) | 99.1 (94.9; 100.0) | 48.2 (11.0; 87.5) | 93.7 (93.3; 94.1) | 92.9 (88.6; 96.0) |
| Early childhood onset § | 56/104 (53.8) | 30/107 (28.0) | 2.99 (1.69; 5.30) | 53.9 (43.8; 63.7) | 72.0 (62.5; 80.2) | 12.6 (9.2; 17.1) | 95.4 (94.2; 96.34) | 70.7 (64.1; 76.7) |
| Fever | 86 (72.3) | 87 (73.1) | 0.96 (0.54; 1.70) | 72.3 (63.3; 80.1) | 26.9 (19.2; 35.8) | 6.9 (6.0; 8.0) | 92.8 (89.5; 95.12) | 30.1 (24.3; 36.30) |
| Crisis CRP elevation | 102 (85.7) | 98 (82.4) | 1.29 (0.64; 2.58) | 85.7 (78.1; 91.5) | 17.7 (11.3; 25.7) | 7.3 (6.6; 8.1) | 94.3 (90.1; 96.72) | 22.4 (17.3; 28.3) |
| Deafness | 19 (16.0) | 2 (1.7) | 11.12 (2.53; 48.89) | 16.0 (9.9; 23.8) | 98.3 (94.1; 99.8) | 41.7 (14.6; 75.0) | 94.0 (93.8; 94.4) | 92.6 (88.5; 95.6) |
| Failure to thrive | 24 (20.2) | 12 (10.1) | 2.25 (1.07; 4.75) | 20.2 (13.4; 28.5) | 89.9 (83.1; 94.7) | 13.1 (7.3; 22.3) | 93.7 (93.1; 94.3) | 85.0 (79.9; 89.3) |
| Pharyngitis | 14 (11.8) | 26 (21.8) | 0.48 (0.24; 0.97) | 11.8 (6.6; 19.0) | 78.2 (69.7; 85.2) | 3.9 (2.2; 6.9) | 92.2 (91.3; 93.0) | 73.5 (67.4; 79.0) |
| Hepatosplenomegaly | 14 (11.8) | 5 (4.2) | 3.04 (1.06; 8.73) | 11.8 (6.6; 19.0) | 95.8 (90.5; 98.6) | 17.4 (7.3; 36.2) | 93.5 (93.0; 94.0) | 89.9 (85.4; 93.4) |
| Characteristics | 32-Gene Panel Subgroup | 55-Gene Panel Subgroup | 62-Gene Panel Subgroup | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cases, n (%) | Controls, n (%) | p | Cases, n (%) | Controls, n (%) | p | Cases, n (%) | Controls, n (%) | p | |||||
| Significantly associated variables | |||||||||||||
| neonatal onset § | 0/4 (0.0) | 1/5 (20.0) | 1 | 1/14 (7.1) | 0/17 (0.0) | 0.45 | 8/49 (16.3) | 0/49 (0.0) | 0.006 | ||||
| early childhood onset § | 2/4 (50.0) | 2/5 (40.0) | 1 | 7/14 (50.0) | 7/17 (41.2) | 0.62 | 30/49 (61.2) | 17/49 (34.7) | 0.009 | ||||
| deafness | 1 (20.0) | 0 (0.0) | 1 | 5 (29.4) | 1 (5.9) | 0.17 | 8 (14.0) | 1 (1.8) | 0.03 | ||||
| failure to thrive | 2 (40.0) | 0 (0.0) | 0.44 | 2 (11.8) | 3 (17.6) | 1 | 14 (24.6) | 7 (12.3) | 0.09 | ||||
| pharyngitis | 0 (0.0) | 2 (40.0) | 0.44 | 3 (17.6) | 4 (23.5) | 1 | 7 (12.3) | 16 (28.1) | 0.04 | ||||
| hepatosplenomegaly | 1 (20.0) | 1 (20.0) | 1 | 4 (23.5) | 0 (0.0) | 0.10 | 7 (12.3) | 1 (1.8) | 0.06 | ||||
| Trend variables | |||||||||||||
| childhood onset § | 4/4 (100.0) | 4/5 (80.0) | 1 | 13/14 (92.9) | 17/17 (1.0) | 0.26 | 45/49 (91.8) | 40/49 (81.6) | 0.14 | ||||
| adulthood onset § | 0/4 (0.0) | 1/5 (20.0) | 1 | 1/14 (7.1) | 0/17 (0.0) | 0.45 | 4/49 (8.2) | 9/49 (18.4) | 0.14 | ||||
| papillitis | 1 (20.0) | 0 (0.0) | 1 | 2 (11.8) | 0 (0.0) | 0.48 | 1 (1.8) | 0 (0.0) | 1 | ||||
| arthritis | 3 (60.0) | 0 (0.0) | 0.17 | 7 (41.2) | 5 (29.4) | 0,47 | 20 (35,1) | 16 (28.1) | 0.42 | ||||
| bipolar aphthosis | 1 (20.0) | 0 (0.0) | 1 | 0 (0.0) | 2 (11.8) | 0.48 | 1 (1.8) | 8 (14.0) | 0.03 | ||||
| Characteristics | 114-Gene Panel Subgroup | 302-Gene Panel Subgroup | |||||||||||
| Cases, n (%) | Controls, n (%) | p | Cases, n (%) | Controls, n (%) | p | ||||||||
| Significantly associated variables | |||||||||||||
| neonatal onset § | 1/9 (11.1) | 0/6 (0.0) | 1 | 2/28 (7.1) | 0/30 (0.0) | 0.23 | |||||||
| early childhood onset § | 5/9 (55.6) | 1/6 (16.7) | 0.29 | 12/28 (42.9) | 6/30 (20.0) | 0.06 | |||||||
| deafness | 1 (11.1) | 0 (0.0) | 1 | 4 (12.9) | 0 (0.0) | 0.11 | |||||||
| failure to thrive | 1 (11.1) | 0 (0.0) | 1 | 5 (16.1) | 2 (6.5) | 0.42 | |||||||
| pharyngitis | 1 (11.1) | 1 (11.1) | 1 | 3 (9.7) | 3 (9.7) | 1 | |||||||
| hepatosplenomegaly | 0 (0.0) | 2 (22.2) | 0.47 | 2 (6.5) | 2 (6.5) | 1 | |||||||
| Trend variables | |||||||||||||
| childhood onset § | 8/9 (88.9) | 4/6 (66.7) | 0.53 | 24/28 (85.7) | 22/30 (73.3) | 0.24 | |||||||
| adulthood onset § | 1/9 (11.1) | 2/6 (33.3) | 0.53 | 4/28 (14.3) | 8/30 (26.7) | 0.24 | |||||||
| papillitis | 0 (0.0) | 0 (0;0) | 1 | 0 (0.0) | 0 (0.0) | 1 | |||||||
| arthritis | 3 (33.3) | 1 (11.1) | 0.58 | 9 (29.0) | 7 (22.6) | 0.56 | |||||||
| bipolar aphthosis | 1 (11.1) | 1 (11.1) | 1 | 5 (16.1) | 6 (19.4) | 0.74 | |||||||
| Characteristics | Cases, n (%) | Controls, n (%) | OR (95% CI) | p-Value |
|---|---|---|---|---|
| Fever § | 86/110 (78.2) | 84/116 (72.4) | 1.37 (0.74; 2.51) | 0.32 |
| Crisis CRP elevation § | 102/105 (97.1) | 92/113 (81.4) | 7.76 (2.24; 26.88) | 0.002 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heiser, L.; Broly, M.; Rittore, C.; Touitou, I.; Georgin-Lavialle, S.; Boursier, G. Predictive Clinical and Biological Criteria for Gene Panel Positivity in Suspected Inherited Autoinflammatory Diseases: Insights from a Case–Control Study. Genes 2023, 14, 1939. https://doi.org/10.3390/genes14101939
Heiser L, Broly M, Rittore C, Touitou I, Georgin-Lavialle S, Boursier G. Predictive Clinical and Biological Criteria for Gene Panel Positivity in Suspected Inherited Autoinflammatory Diseases: Insights from a Case–Control Study. Genes. 2023; 14(10):1939. https://doi.org/10.3390/genes14101939
Chicago/Turabian StyleHeiser, Lionel, Martin Broly, Cécile Rittore, Isabelle Touitou, Sophie Georgin-Lavialle, and Guilaine Boursier. 2023. "Predictive Clinical and Biological Criteria for Gene Panel Positivity in Suspected Inherited Autoinflammatory Diseases: Insights from a Case–Control Study" Genes 14, no. 10: 1939. https://doi.org/10.3390/genes14101939
APA StyleHeiser, L., Broly, M., Rittore, C., Touitou, I., Georgin-Lavialle, S., & Boursier, G. (2023). Predictive Clinical and Biological Criteria for Gene Panel Positivity in Suspected Inherited Autoinflammatory Diseases: Insights from a Case–Control Study. Genes, 14(10), 1939. https://doi.org/10.3390/genes14101939