
Usnic Acid Derivatives Inhibit DNA Repair Enzymes Tyrosyl-DNA Phosphodiesterases 1 and 2 and Act as Potential Anticancer Agents
 , , , , , ,
, , , , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Gel-Based Tdp2 Activity Assay
2.2. Cytotoxicity Assay
2.3. Tdp2 Knockout HEK293FT Cell Clones
2.4. Molecular Modeling
3. Results
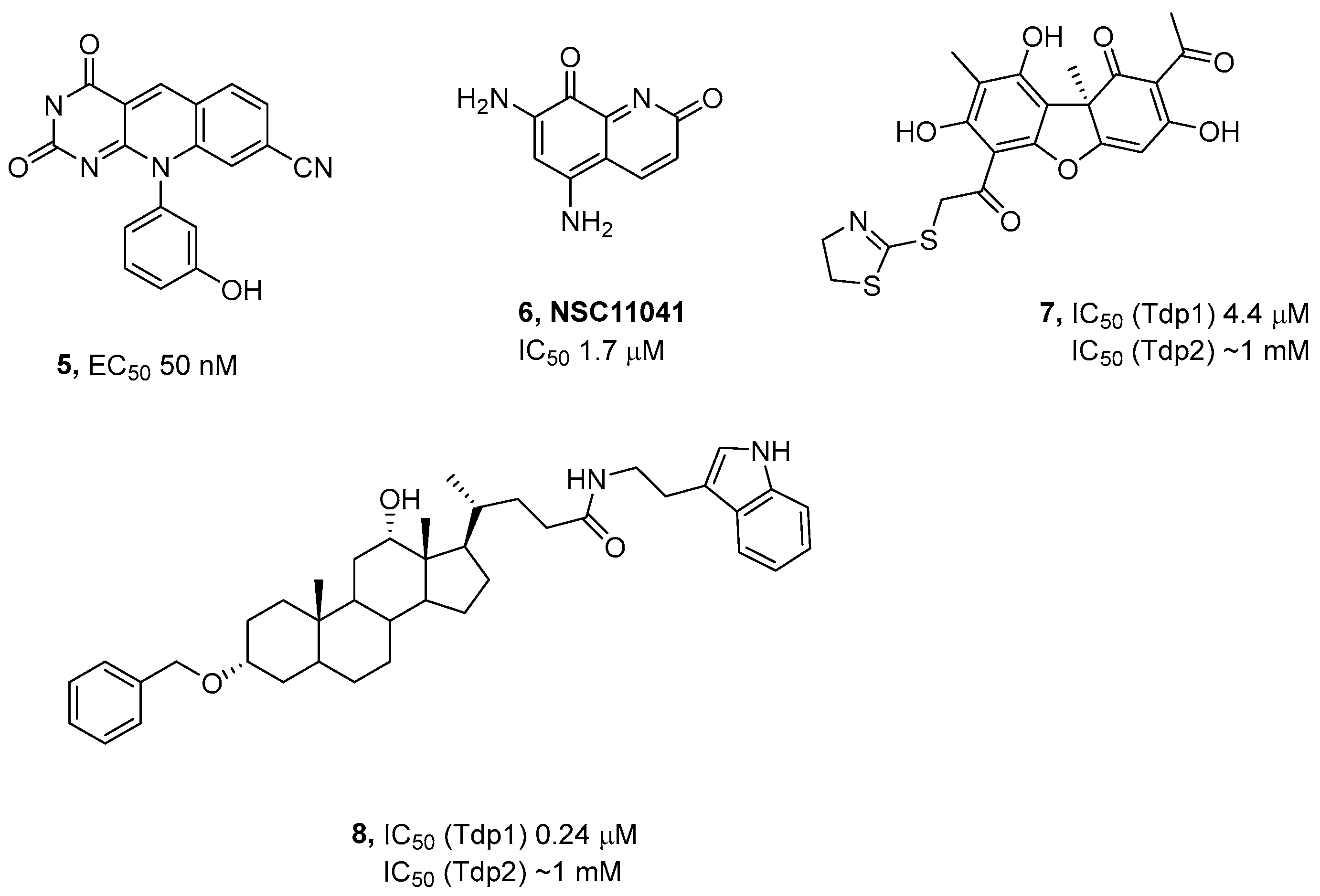
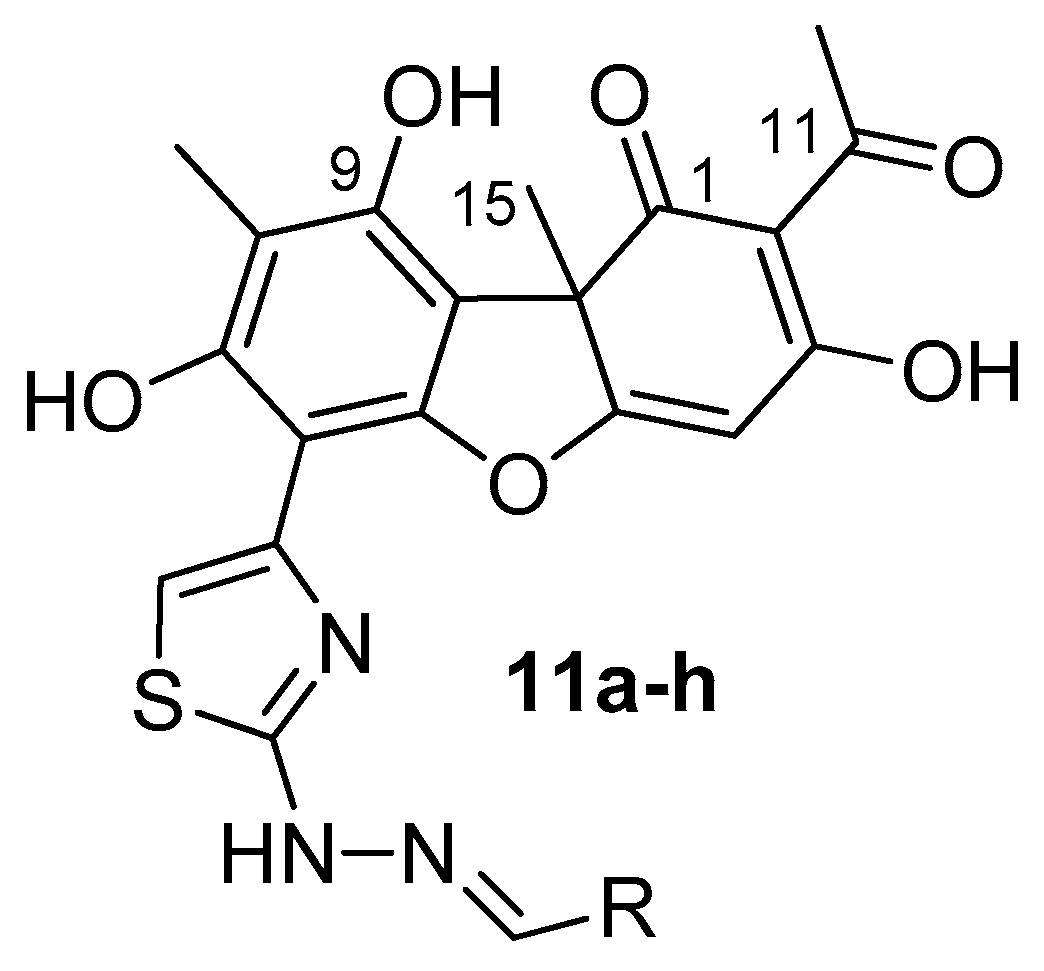
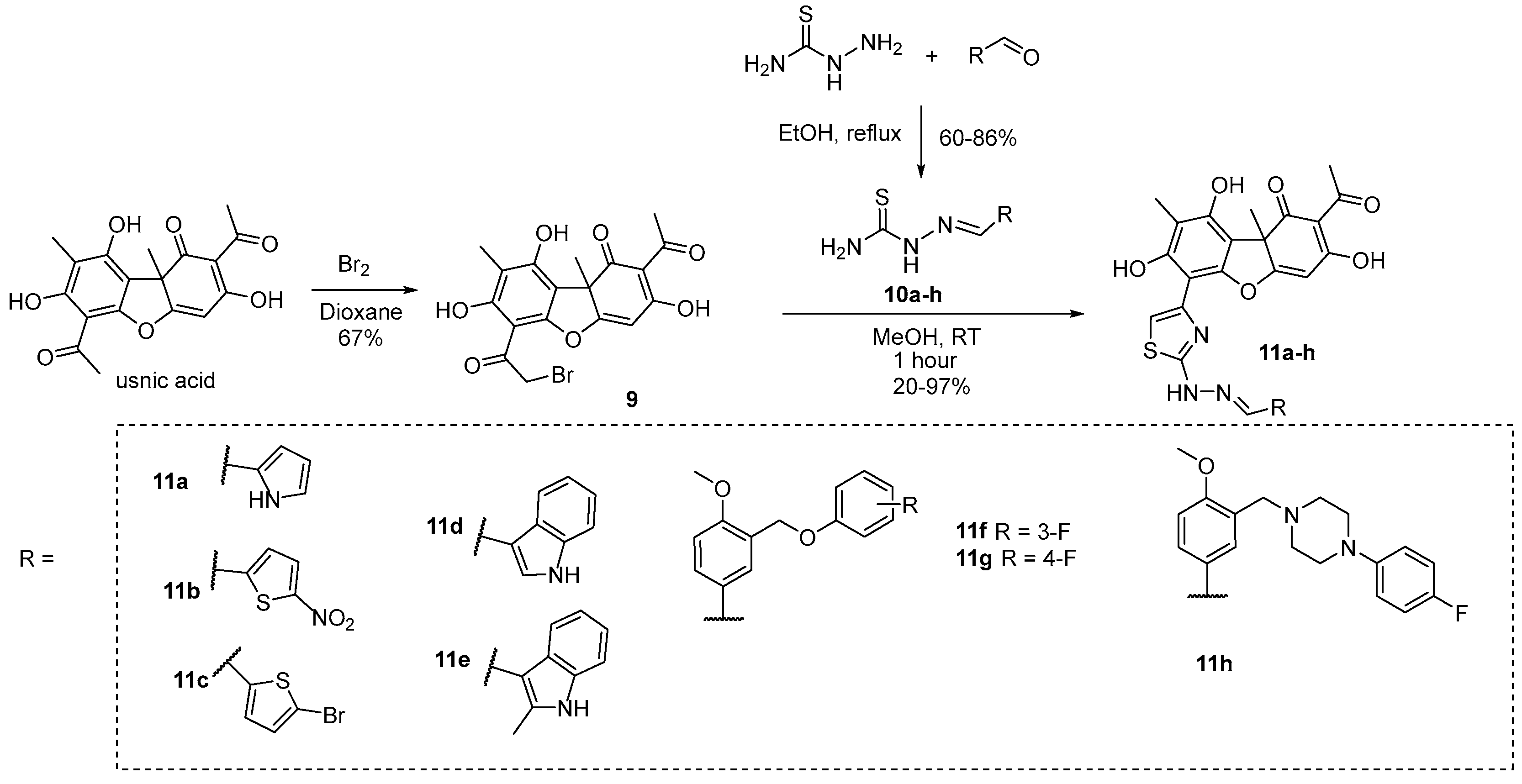
3.1. Chemistry
3.2. Effect of Compounds on the Activity of Purified Tdp2
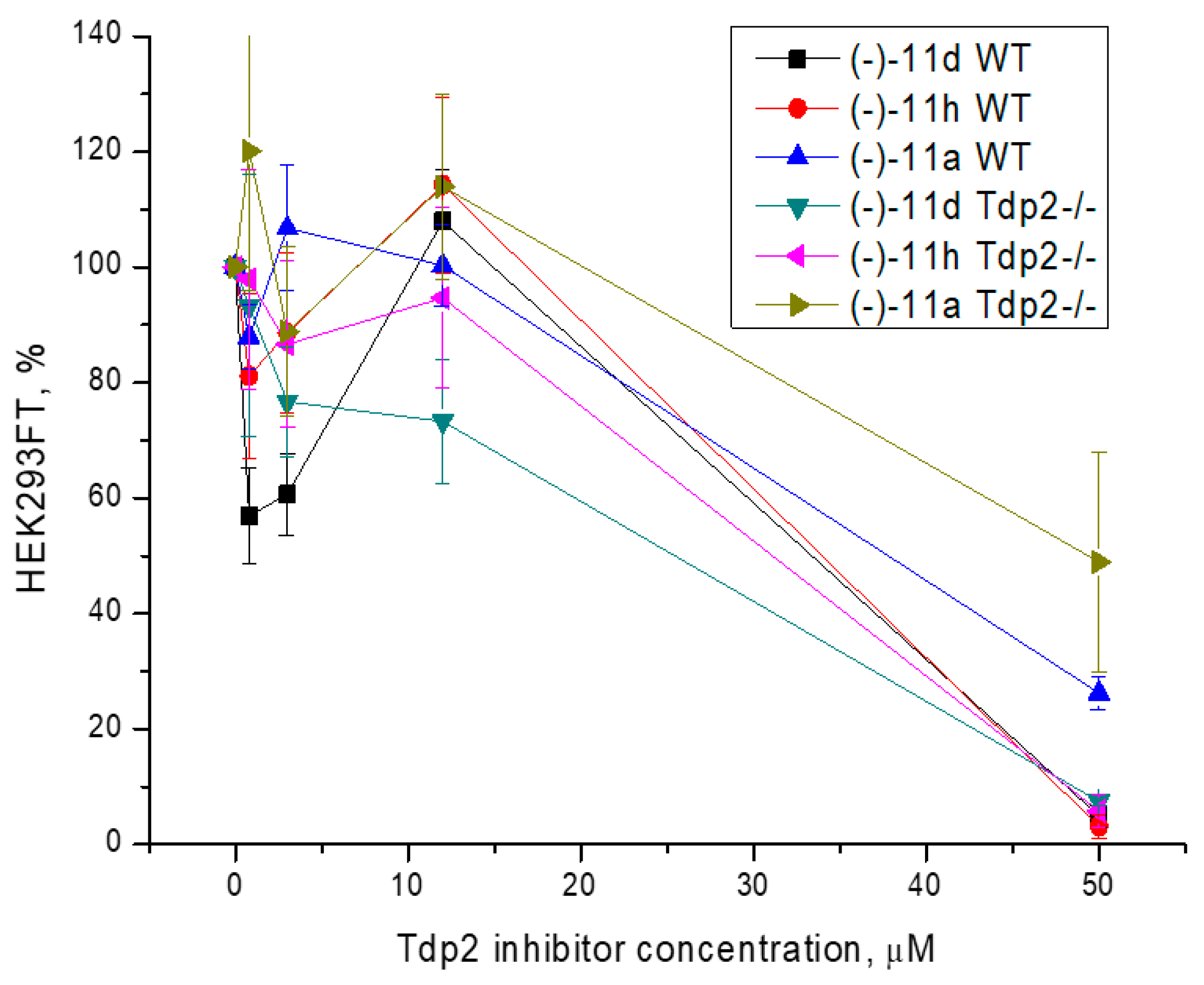
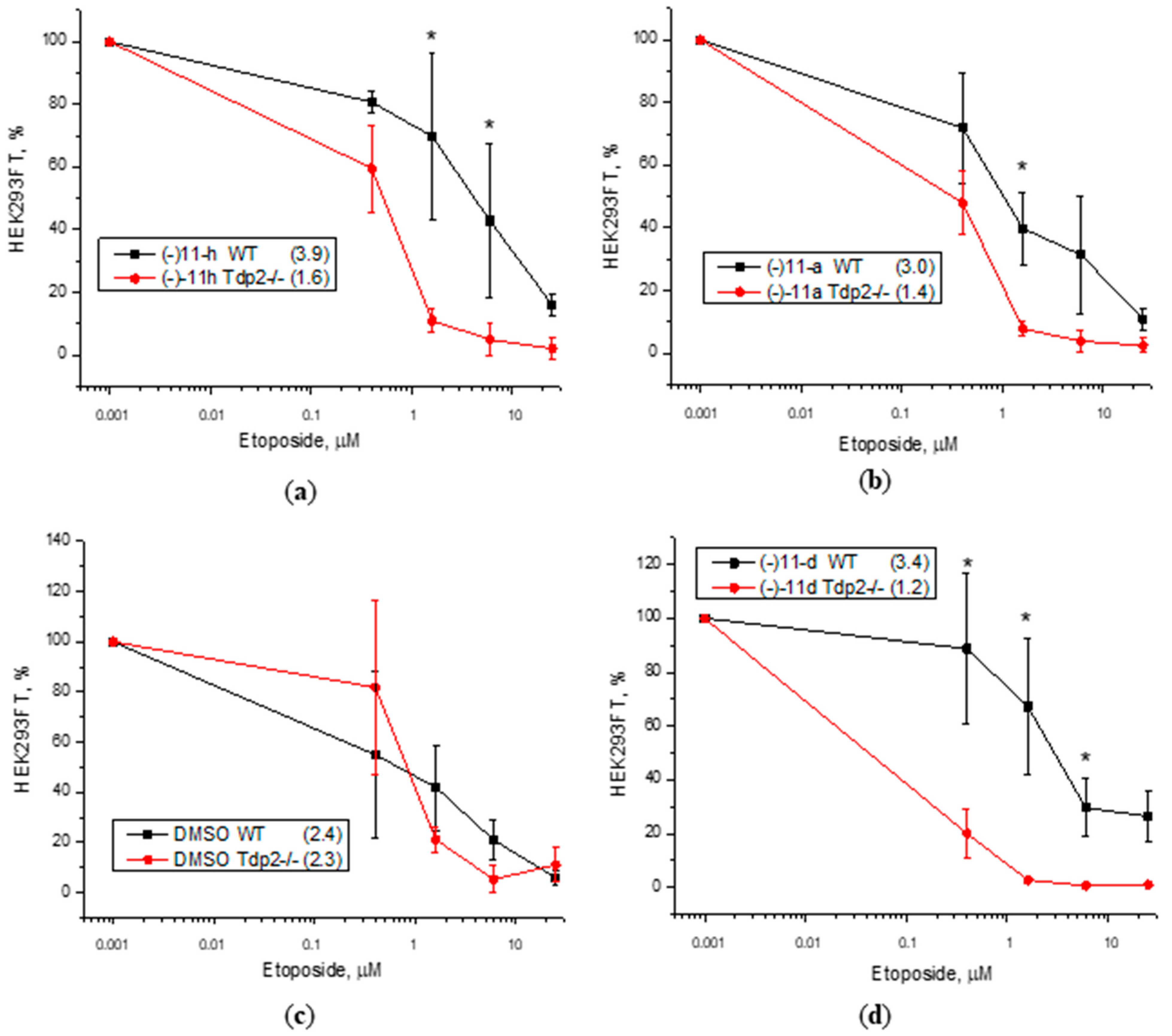
3.3. Cytotoxicity Assay
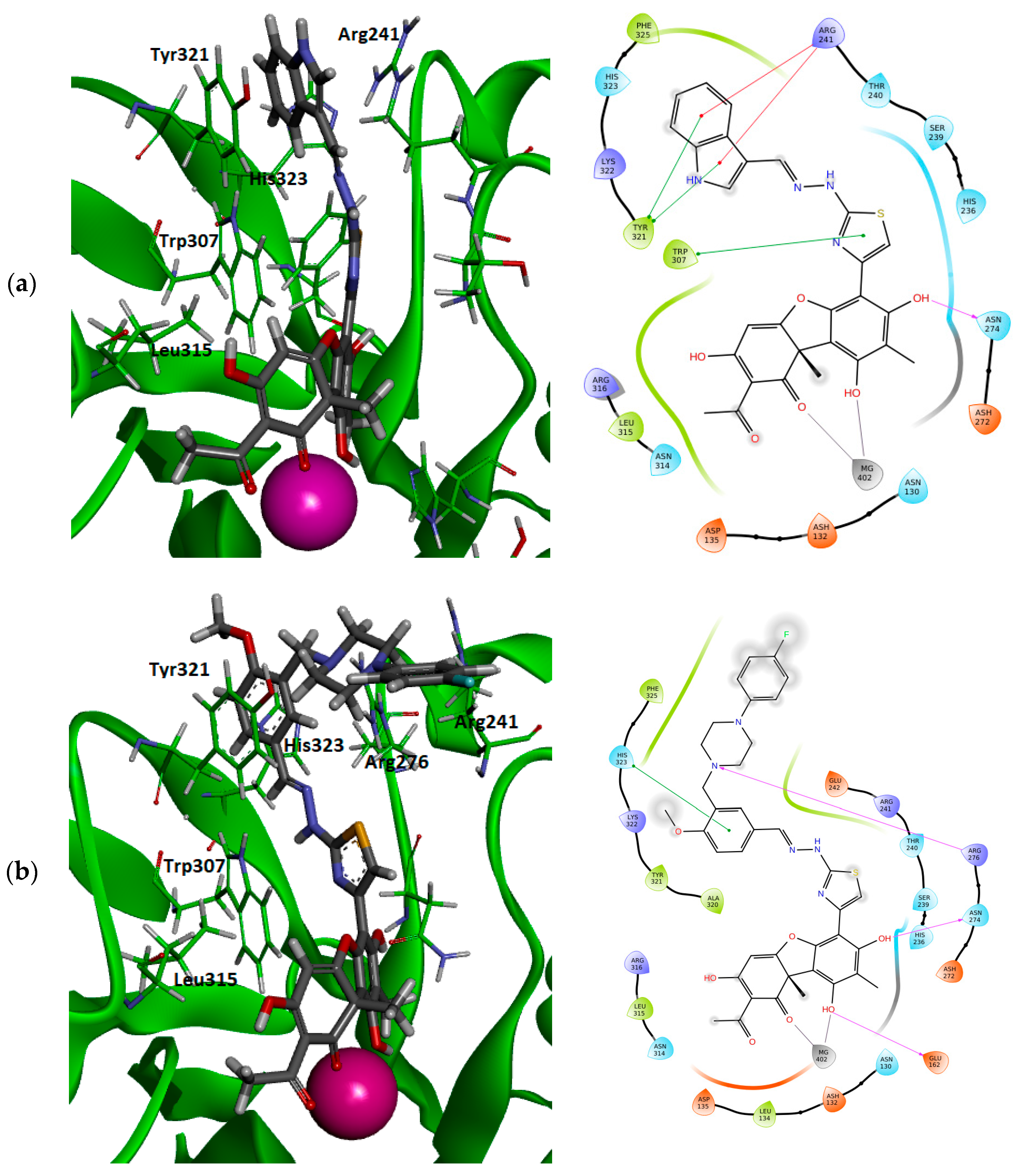
3.4. Molecular Modeling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kelley, M.R.; Logsdon, D.; Fishel, M.L. Targeting DNA repair pathways for cancer treatment: What’s new? Future Oncol. 2014, 10, 1215–1237. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. Inhibiting the DNA damage response as a therapeutic manoeuvre in cancer. Br. J. Pharmacol. 2013, 169, 1745–1765. [Google Scholar] [CrossRef] [PubMed]
- Staker, B.L.; Hjerrild, K.; Feese, M.D.; Behnke, C.A.; Burgin, A.B., Jr.; Stewart, L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. USA 2002, 99, 15387–15392. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef]
- Pommier, Y.; Huang, S.Y.; Gao, R.; Das, B.B.; Murai, J.; Marchand, C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair 2014, 19, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Interthal, H.; Pouliott, J.J.; Champoux, J.J. The Tyrosyl-DNA Phosphodiesterase Tdp1 Is a Member of the Phospholipase D Superfamily. Proc. Natl. Acad. Sci. USA 2001, 98, 12009–12014. [Google Scholar] [CrossRef]
- Ben Hassine, S.; Arcangioli, B. Tdp1 protects against oxidative DNA damage in non-dividing fission yeast. EMBO J. 2009, 28, 632–640. [Google Scholar] [CrossRef]
- Povirk, L.F. Processing of damaged DNA ends for double-strand break repair in mammalian cells. ISRN Mol. Biol. 2012, 2012, 345805. [Google Scholar] [CrossRef]
- Comeaux, E.Q.; Van Waardenburg, R.C.A.M. Tyrosyl-DNA Phosphodiesterase I Resolves Both Naturally and Chemically Induced DNA Adducts and Its Potential as a Therapeutic Target. Drug Metab. Rev. 2014, 46, 494–507. [Google Scholar] [CrossRef]
- Brettrager, E.J.; van Waardenburg, R.C.A.M. Targeting Tyrosyl-DNA phosphodiesterase I to enhance toxicity of phosphodiester linked DNA-adducts. Cancer Drug Resist. 2019, 2, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Hirano, R.; Interthal, H.; Huang, C.; Nakamura, T.; Deguchi, K.; Choi, K.; Bhattacharjee, M.; Arimura, K.; Umehara, F.; Izumo, S.; et al. Spinocerebellar ataxia with axonal neuropathy: Consequence of a Tdp1 recessive neomorphic mutation? EMBO J. 2007, 26, 4732–4743. [Google Scholar] [CrossRef]
- He, X.; van Waardenburg, R.C.A.M.; Babaoglu, K.; Price, A.C.; Nitiss, K.C.; Nitiss, J.L.; Bjornsti, M.-A.; White, S.W. Mutation of a conserved active site residue converts tyrosyl-DNA phosphodiesterase I into a DNA topoisomerase I-dependent poison. J. Mol. Biol. 2007, 372, 1070–1081. [Google Scholar] [CrossRef] [PubMed]
- Interthal, H.; Chen, H.J.; Kehl-Fie, T.E.; Zotzmann, J.; Leppard, J.B.; Champoux, J.J. SCAN1 mutant Tdp1 accumulates the enzyme–DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 2005, 24, 2224–2233. [Google Scholar] [CrossRef] [PubMed]
- Katyal, S.; Khamisy, S.F.; Russell, H.R.; Li, Y.; Ju, L.; Caldecott, K.W.; McKinnon, P.J. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 2007, 26, 4720–4731. [Google Scholar] [CrossRef]
- El-Khamisy, S.F.; Katyal, S.; Patel, P.; Ju, L.; McKinnon, P.J.; Caldecott, K.W. Synergistic decrease of DNA single-strand break repair rates in mouse neural cells lacking both Tdp1 and aprataxin. DNA Repair 2009, 8, 760–766. [Google Scholar] [CrossRef]
- Barthelmes, H.U.; Habermeyer, M.; Christensen, M.O.; Mielke, C.; Interthal, H.; Pouliot, J.J.; Boege, F.; Marko, D. TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II. J. Biol. Chem. 2004, 279, 55618–55625. [Google Scholar] [CrossRef]
- Nivens, M.C.; Felder, T.; Galloway, A.H.; Pena, M.M.; Pouliot, J.J.; Spencer, H.T. Engineered resistance to camptothecin and antifolates by retroviral coexpression of tyrosyl DNA phosphodiesterase-I and thymidylate synthase. Cancer Chemother. Pharmacol. 2004, 53, 107–115. [Google Scholar] [CrossRef]
- Alagoz, M.; Gilbert, D.C.; El-Khamisy, S.; Chalmers, A.J. DNA repair and resistance to topoisomerase I inhibitors: Mechanisms, biomarkers and therapeutic targets. Curr. Med. Chem. 2012, 19, 3874–3885. [Google Scholar] [CrossRef]
- Perego, P.; Cossa, G.; Tinelli, S.; Corna, E.; Carenini, N.; Gatti, L.; De Cesare, M.; Ciusani, E.; Zunino, F.; Luison, E.; et al. Role of tyrosyl-DNA phosphodiesterase 1 and inter-players in regulation of tumor cell sensitivity to topoisomerase I inhibition. Biochem. Pharmacol. 2012, 83, 27–36. [Google Scholar] [CrossRef]
- Meisenberg, C.; Ward, S.E.; Schmid, P.; El-Khamisy, S.F. TDP1/TOP1 ratio as a promising indicator for the response of small cell lung cancer to topotecan. J. Cancer Sci. Ther. 2014, 6, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Zakharenko, A.L.; Luzina, O.A.; Chepanova, A.A.; Dyrkheeva, N.S.; Salakhutdinov, N.F.; Lavrik, O.I. Natural Products and Their Derivatives as Inhibitors of the DNA Repair Enzyme Tyrosyl-DNA Phosphodiesterase 1. Int. J. Mol. Sci. 2023, 24, 5781. [Google Scholar] [CrossRef]
- Zeng, Z.; Cortés-Ledesma, F.; El Khamisy, S.F.; Caldecott, K.W. TDP2/TTRAP is the major 5′-tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II-induced DNA damage. J. Biol. Chem. 2011, 286, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Ledesma, F.C.; El Khamisy, S.F.; Zuma, M.C.; Osborn, K.; Caldecott, K.W. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 2009, 461, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Herreros, F.; Romero-Granados, R.; Zeng, Z.; Alvarez-Quilón, A.; Quintero, C.; Ju, L.; Umans, L.; Vermeire, L.; Huylebroeck, D.; Caldecott, K.W.; et al. TDP2-dependent non-homologous end-joining protects against topoisomerase II-induced DNA breaks and genome instability in cells and in vivo. PLoS Genet. 2013, 9, e1003226. [Google Scholar] [CrossRef]
- Zakharenko, A.; Dyrkheeva, N.; Lavrik, O. Dual DNA topoisomerase 1 and tyrosyl-DNA phosphodiesterase 1 inhibition for improved anticancer activity. Med. Res. Rev. 2019, 39, 1427–1441. [Google Scholar] [CrossRef]
- Kawale, A.S.; Povirk, L.F. Tyrosyl-DNA phosphodiesterases: Rescuing the genome from the risks of relaxation. Nucleic Acids Res. 2018, 46, 520–537. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Dexheimer, T.S.; Takeda, S.; Pommier, Y. Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. J. Biol. Chem. 2012, 287, 12848–12857. [Google Scholar] [CrossRef]
- Zeng, Z.; Sharma, A.; Ju, L.; Murai, J.; Umans, L.; Vermeire, L.; Pommier, Y.; Takeda, S.; Huylebroeck, D.; Caldecott, K.W.; et al. TDP2 promotes repair of topoisomerase I-mediated DNA damage in the absence of TDP1. Nucleic Acids Res. 2012, 40, 8371–8380. [Google Scholar] [CrossRef]
- Maede, Y.; Shimizu, H.; Fukushima, T.; Kogame, T.; Nakamura, T.; Miki, T.; Takeda, S.; Pommier, Y.; Murai, J. Differential and common DNA repair pathways for topoisomerase I- and II-targeted drugs in a genetic DT40 repair cell screen panel. Mol. Cancer Ther. 2014, 13, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Zakharenko, A.L.; Luzina, O.A.; Sokolov, D.N.; Kaledin, V.I.; Nikolin, V.P.; Popova, N.A.; Patel, J.; Zakharova, O.D.; Chepanova, A.A.; Zafar, A.; et al. Novel tyrosyl-DNA phosphodiesterase 1 inhibitors enhance the therapeutic impact of topotecan on in vivo tumor models. Eur. J. Med. Chem. 2019, 161, 581–593. [Google Scholar] [CrossRef]
- Khomenko, T.M.; Zakharenko, A.L.; Chepanova, A.A.; Ilina, E.S.; Zakharova, O.D.; Kaledin, V.I.; Nikolin, V.P.; Popova, N.A.; Korchagina, D.V.; Reynisson, J.; et al. Promising New Inhibitors of Tyrosyl-DNA Phosphodiesterase I (Tdp 1) Combining 4-Arylcoumarin and Monoterpenoid Moieties as Components of Complex Antitumor Therapy. Int. J. Mol. Sci. 2019, 21, 126. [Google Scholar] [CrossRef] [PubMed]
- Nikolin, V.P.; Popova, N.A.; Kaledin, V.I.; Luzina, O.A.; Zakharenko, A.L.; Salakhutdinov, N.F.; Lavrik, O.I. The influence of an enamine usnic acid derivative (a tyrosyl-DNA phosphodiesterase 1 inhibitor) on the therapeutic effect of topotecan against transplanted tumors in vivo. Clin. Exp. Metastasis 2021, 38, 431–440. [Google Scholar] [CrossRef]
- Chernyshova, I.A.; Zakharenko, A.L.; Kurochkin, N.N.; Dyrkheeva, N.S.; Kornienko, T.E.; Popova, N.A.; Nikolin, V.P.; Ilina, E.S.; Zharkov, T.D.; Kupryushkin, M.S.; et al. Lipophilic Purine Nucleoside-Tdp1 Inhibitor-Enhances DNA Damage Induced by Topotecan In Vitro and Potentiates the Antitumor Effect of Topotecan In Vivo. Molecules 2022, 28, 323. [Google Scholar] [CrossRef]
- Marchand, C.; Abdelmalak, M.; Kankanala, J.; Huang, S.Y.; Kiselev, E.; Fesen, K.; Kurahashi, K.; Sasanuma, H.; Takeda, S.; Aihara, H.; et al. Deazaflavin Inhibitors of Tyrosyl-DNA Phosphodiesterase 2 (TDP2) Specific for the Human Enzyme and Active against Cellular TDP2. ACS Chem. Biol. 2016, 11, 1925–1933. [Google Scholar] [CrossRef]
- Kankanala, J.; Ribeiro, C.J.A.; Kiselev, E.; Ravji, A.; Williams, J.; Xie, J.; Aihara, H.; Pommier, Y.; Wang, Z. Novel Deazaflavin Analogues Potently Inhibited Tyrosyl DNA Phosphodiesterase 2 (TDP2) and Strongly Sensitized Cancer Cells toward Treatment with Topoisomerase II (TOP2) Poison Etoposide. J. Med. Chem. 2019, 9, 4669–4682. [Google Scholar] [CrossRef]
- Yu, L.M.; Hu, Z.; Chen, Y.; Ravji, A.; Lopez, S.; Plescia, C.B.; Yu, Q.; Yang, H.; Abdelmalak, M.; Saha, S.; et al. Synthesis and structure-activity relationship of furoquinolinediones as inhibitors of Tyrosyl-DNA phosphodiesterase 2 (TDP2). Eur. J. Med. Chem. 2018, 10, 777–796. [Google Scholar] [CrossRef] [PubMed]
- Kont, Y.S.; Dutta, A.; Mallisetty, A.; Mathew, J.; Minas, T.; Kraus, C.; Dhopeshwarkar, P.; Kallakury, B.; Mitra, S.; Üren, A.; et al. Depletion of tyrosyl DNA phosphodiesterase 2 activity enhances etoposide-mediated double-strand break formation and cell killing. DNA Repair 2016, 43, 38–47. [Google Scholar] [CrossRef]
- Dyrkheeva, N.S.; Filimonov, A.S.; Luzina, O.A.; Orlova, K.A.; Chernyshova, I.A.; Kornienko, T.E.; Malakhova, A.A.; Medvedev, S.P.; Zakharenko, A.L.; Ilina, E.S.; et al. New Hybrid Compounds Combining Fragments of Usnic Acid and Thioether Are Inhibitors of Human Enzymes TDP1, TDP2 and PARP1. Int. J. Mol. Sci. 2021, 22, 11336. [Google Scholar] [CrossRef]
- Salomatina, O.V.; Dyrkheeva, N.S.; Popadyuk, I.I.; Zakharenko, A.L.; Ilina, E.S.; Komarova, N.I.; Reynisson, J.; Salakhutdinov, N.F.; Lavrik, O.I.; Volcho, K.P. New Deoxycholic Acid Derived Tyrosyl-DNA Phosphodiesterase 1 Inhibitors Also Inhibit Tyrosyl-DNA Phosphodiesterase 2. Molecules 2021, 27, 72. [Google Scholar] [CrossRef] [PubMed]
- Luzina, O.A.; Salakhutdinov, N.F. Biological activity of usnic acid and its derivatives: Part 1. Activity against unicellular organisms. Russ. J. Bioorg. Chem. 2016, 42, 115–132. [Google Scholar] [CrossRef]
- Luzina, O.A.; Salakhutdinov, N.F. Biological activity of usnic acid and its derivatives: Part 2. effects on higher organisms. Molecular and physicochemical aspects. Russ. J. Bioorg. Chem. 2016, 42, 249–268. [Google Scholar] [CrossRef]
- Filimonov, A.S.; Chepanova, A.A.; Luzina, O.A.; Zakharenko, A.L.; Zakharova, O.D.; Ilina, E.S.; Dyrkheeva, N.S.; Kuprushkin, M.S.; Kolotaev, A.V.; Khachatryan, D.S.; et al. New Hydrazinothiazole Derivatives of Usnic Acid as Potent Tdp1 Inhibitors. Molecules 2019, 24, 3711. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.J. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Schellenberg, M.J.; Appel, C.D.; Adhikari, S.; Robertson, P.D.; Ramsden, D.A.; Williams, S. Mechanism of repair of 5′-topoisomerase II–DNA adducts by mammalian tyrosyl-DNA phosphodiesterase 2. Nat. Struct. Mol. Biol. 2012, 19, 1363–1371. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Polovinka, M.P.; Salakhutdinov, N.F.; Panchenko, M.Y. Method for Preparing Usninic Acid. Patent RU2317076, 20 February 2008. [Google Scholar]
- Luzina, O.A.; Sokolov, D.N.; Shernyukov, A.V.; Salakhutdinov, N.F. Synthesis of aurones based on usninic acid. Chem. Nat. Compd. 2012, 48, 385–391. [Google Scholar] [CrossRef]
- Riccio, A.A.; Schellenberg, M.J.; Williams, R.S. Molecular mechanisms of topoisomerase 2 DNA-protein crosslink resolution. Cell. Mol. Life Sci. 2020, 77, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, M.J.; Perera, L.; Strom, C.N.; Waters, C.A.; Monian, B.; Appel, C.D.; Vilas, C.K.; Williams, J.G.; Ramsden, D.A.; Williams, R.S. Reversal of DNA damage induced Topoisomerase 2 DNA–protein crosslinks by Tdp2. Nucleic Acids Res. 2016, 44, 3829–3844. [Google Scholar] [CrossRef] [PubMed]
- Zakharenko, A.; Luzina, O.; Koval, O.; Nilov, D.; Gushchina, I.; Dyrkheeva, N.; Švedas, V.; Salakhutdinov, N.; Lavrik, O. Tyrosyl-DNA Phosphodiesterase 1 Inhibitors: Usnic Acid Enamines Enhance the Cytotoxic Effect of Camptothecin. J. Nat. Prod. 2016, 79, 2961–2967. [Google Scholar] [CrossRef]
- Dyrkheeva, N.; Luzina, O.; Filimonov, A.; Zakharova, O.; Ilina, E.; Zakharenko, A.; Kuprushkin, M.; Nilov, D.; Gushchina, I.; Švedas, V.; et al. Inhibitory Effect of New Semisynthetic Usnic Acid Derivatives on Human Tyrosyl-DNA Phosphodiesterase 1. Planta Med. 2019, 85, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Galanty, A.; Pasko, P.; Podolak, I. Enantioselective activity of usnic acid: A comprehensive review and future perspectives. Phytochem. Rev. 2019, 18, 527–548. [Google Scholar] [CrossRef]
- Macedo, D.C.S.; Almeida, F.J.F.; Wanderley, M.S.O.; Ferraz, M.S.; Santos, N.P.S.; López, A.M.Q.; Santos-Magalhães, N.S.; Lira-Nogueira, M.C.B. Usnic acid: From an ancient lichen derivative to promising biological and nanotechnology applications. Phytochem. Rev. 2021, 20, 609–630. [Google Scholar] [CrossRef]
- Wang, P.; Elsayed, M.S.A.; Plescia, C.B.; Ravji, A.; Redon, C.E.; Kiselev, E.; Marchand, C.; Zeleznik, O.; Agama, K.; Pommier, Y.; et al. Synthesis and Biological Evaluation of the First Triple Inhibitors of Human Topoisomerase 1, Tyrosyl-DNA Phosphodiesterase 1 (Tdp1), and Tyrosyl-DNA Phosphodiesterase 2 (Tdp2). J. Med. Chem. 2017, 60, 3275–3288. [Google Scholar] [CrossRef]
- Beck, D.E.; Lv, W.; Abdelmalak, M.; Plescia, C.B.; Agama, K.; Marchand, C.; Pommier, Y.; Cushman, M. Synthesis and biological evaluation of new fluorinated and chlorinated indenoisoquinoline topoisomerase I poisons. Bioorg. Med. Chem. 2016, 24, 1469–1479. [Google Scholar] [CrossRef][Green Version]
- Ivankin, D.I.; Dyrkheeva, N.S.; Zakharenko, A.L.; Ilina, E.S.; Zarkov, T.O.; Reynisson, J.; Luzina, O.A.; Volcho, K.P.; Salakhutdinov, N.F.; Lavrik, O.I. Monoterpene substituted thiazolidin-4-ones as novel TDP1 inhibitors: Synthesis, biological evaluation and docking. Bioorg. Med. Chem. Lett. 2022, 73, 128909. [Google Scholar] [CrossRef]
- Khomenko, T.M.; Zakharenko, A.L.; Kornienko, T.E.; Chepanova, A.A.; Dyrkheeva, N.S.; Artemova, A.O.; Korchagina, D.V.; Achara, C.; Curtis, A.; Reynisson, J.; et al. New 5-Hydroxycoumarin-Based Tyrosyl-DNA Phosphodiesterase I Inhibitors Sensitize Tumor Cell Line to Topotecan. Int. J. Mol. Sci. 2023, 24, 9155. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | R |  |  | ||
| Tdp1 * | Tdp2 | Tdp1 * | Tdp2 | ||
| IC50, nM | IC50, µM | IC50, nM | IC50, µM | ||
| 11a |  | 188 ± 3 | >50 | 18 ± 1 | 8 ± 4 |
| (16m) * | |||||
| 11d |  | 26 ± 6 | >50 | 57 ± 1 | 6 ± 3 |
| (16p) * | |||||
| 11h |  | 80 ± 7 | >50 | 59 ± 5 | 9 ± 1 |
| (17i) * | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakharenko, A.L.; Dyrkheeva, N.S.; Luzina, O.A.; Filimonov, A.S.; Mozhaitsev, E.S.; Malakhova, A.A.; Medvedev, S.P.; Zakian, S.M.; Salakhutdinov, N.F.; Lavrik, O.I. Usnic Acid Derivatives Inhibit DNA Repair Enzymes Tyrosyl-DNA Phosphodiesterases 1 and 2 and Act as Potential Anticancer Agents. Genes 2023, 14, 1931. https://doi.org/10.3390/genes14101931
Zakharenko AL, Dyrkheeva NS, Luzina OA, Filimonov AS, Mozhaitsev ES, Malakhova AA, Medvedev SP, Zakian SM, Salakhutdinov NF, Lavrik OI. Usnic Acid Derivatives Inhibit DNA Repair Enzymes Tyrosyl-DNA Phosphodiesterases 1 and 2 and Act as Potential Anticancer Agents. Genes. 2023; 14(10):1931. https://doi.org/10.3390/genes14101931
Chicago/Turabian StyleZakharenko, Alexandra L., Nadezhda S. Dyrkheeva, Olga A. Luzina, Aleksandr S. Filimonov, Evgenii S. Mozhaitsev, Anastasia A. Malakhova, Sergey P. Medvedev, Suren M. Zakian, Nariman F. Salakhutdinov, and Olga I. Lavrik. 2023. "Usnic Acid Derivatives Inhibit DNA Repair Enzymes Tyrosyl-DNA Phosphodiesterases 1 and 2 and Act as Potential Anticancer Agents" Genes 14, no. 10: 1931. https://doi.org/10.3390/genes14101931
APA StyleZakharenko, A. L., Dyrkheeva, N. S., Luzina, O. A., Filimonov, A. S., Mozhaitsev, E. S., Malakhova, A. A., Medvedev, S. P., Zakian, S. M., Salakhutdinov, N. F., & Lavrik, O. I. (2023). Usnic Acid Derivatives Inhibit DNA Repair Enzymes Tyrosyl-DNA Phosphodiesterases 1 and 2 and Act as Potential Anticancer Agents. Genes, 14(10), 1931. https://doi.org/10.3390/genes14101931