1. Introduction
Since the introduction of the neutral theory [
1,
2], base composition and substitution rates of noncoding sequences have been used as a proxy for neutral evolution. This provides one mechanism to study the role of selection on putatively neutral substitutions within coding regions, particularly silent substitutions and any associated codon usage bias [
1,
3,
4]. Although there is ample evidence for selection on some noncoding sequences [
5,
6], the use of intergenic sequence data to estimate neutral evolution assumes that the underlying mutation process is the same across all sites regardless of any functional property of the site, such as coding for an amino acid sequence, which should only affect the subsequent fixation process through selection.
Some recent work has raised questions about this long-held assumption about mutations. Evidence was reported for a decreased mutation rate in exons relative to introns in human DNA [
4] and, although this was not supported by an analysis of de novo mutations in germline cells [
7], it raises an important question for genome evolution. Although it is not clear what could cause such a difference, it needs to be better established whether or not there is any such relationship between mutation rate and function. Here, I further address this issue using the flowering plant chloroplast genome (cpDNA). Previous studies have shown that substitution dynamics of intergenic, noncoding (NC) DNA in Angiosperm cpDNA are complex. Most notably, they are strongly context-dependent, with evidence for at least three neighboring bases on each side of any site affecting the process, resulting in a wide range of substitution dynamics across sites [
8,
9]. These complex dynamics have been used as an estimate of neutral evolution to show that the codon usage bias (CUB) of chloroplast genes appears to be largely driven by mutation bias [
10].
In the current analysis, substitutions at fourfold degenerate (FFD) sites in protein-coding sequences of Angiosperm cpDNA are analyzed in a context-dependent manner and then compared to what has been observed previously in NC regions. There is no reason to expect a priori that selective constraints across sites are associated with either context or type of mutation (e.g., transition or transversion), so the hypothesis is that variation in context-dependent substitution dynamics is the same across both FFD and NC sites. Any influence of selection is expected to affect only the overall substitution rate, so any other type of variation would be attributable to variation in mutation dynamics. Here, I include repair processes, such as mismatch repair, as part of mutation dynamics, so that mutation includes the phenomena that play out within an individual before the population-level selection process occurs. The data show that FFD and NC sites display different substitution dynamics, while context effects are essentially the same at FFD sites as in NC regions; there are two small but consistent differences between the two types of sites. First, the rate of substitution is greater at FFD sites with the exception of one specific type of substitution: A → T and T → A (W → W) transversions. Second, the predicted equilibrium A+T content is higher at FFD sites in a majority of contexts, and this is matched by observed compositional differences.
Possible neutral explanations for this difference between FFD and NC sites are explored. One is that there is an influence of transcription on mutations, and a second is that there are subtle context effects from a broader neighboring region than the tetranucleotide analyzed in this study, which could be causing the observed differences in substitution as a result of average context differences between FFD and NC sites. Although transcription appears to have some effect on substitution rate, the data do not support either of these two mechanisms explaining the difference between FFD and NC sites. The third possibility is that the mutation process is not quite equivalent at the two site types. This does not require a difference in mutation dynamics resulting directly from function. Rather, it could be a consequence of some other difference that is related to functionality and/or expression. The evidence shows that there is a potential CG effect—an increased rate of C → T substitutions upstream of a G that could arise from a high rate of deamination of methylated Cs—and although it is shown that any CG effect cannot account for the difference between FFD and NC sites, it is proposed that the difference between FFD and NC sites might be due to differences in epigenetic modifications that give rise to variation in mutation dynamics across selectively neutral sites. Regardless of the specific reason, though, the results indicate that there are complications to the application of NC data in analyses of selection on coding sequences.
2. Materials and Methods
The analysis of substitutions followed the methodology described previously for noncoding DNA regions [
9]. RefSeq complete Angiosperm chloroplast genome sequences were downloaded from NCBI
www.ncbi.nlm.nih.gov/genome/browse#!/eukaryotes/ (accessed on 14 March 2019) and then parsed with the Biopython 1.76 [
11]. Genomes were grouped into 280 closely related triplets from 40 different families, which were chosen to ensure non-overlapping lineages, and the coding strand of each CDS was then aligned using the
pairwise function from Biopython with the parameters set as
match = 2,
mismatch = −1,
gap open = −2, and
gap extend = −0.5. Any CDS alignment with more than 30 total gaps introduced was excluded from further analysis.
The alignments were used to generate 128 tetranucleotide context-dependent substitution matrices, one matrix for each of the tetranucleotide contexts within which a FFD site can occur (e.g., no FFD site can have a 5′ A due to the genetic code.) Any substitution site N
0 can be considered as the central site of the pentanucleotide {N
2 N
1 [N
0] N
1 N
2} where N
1, and N
2 are the neighboring base pairs that compose the tetranucleotide context flanking N
0. To determine the codon position of any N
0 site, it was assumed that any gaps introduced during alignment did not change the reading frame, so positions that were a multiple of three were assumed to be potential FFD sites (which is determined by the content of the 5′ N
2 and N
1 nucleotides). Count Matrices were generated and then the complementary matrices combined, following which Rate Matrices (also Markov Transition matrices) and the stationary vector for each matrix were all performed as described previously [
9]. All comparisons were between the combined strand matrices, or the matrices generated by combining complementary pairs. To estimate the sampling error for the A+T content of the stationary vector, a bootstrap method was employed. Each resample generated a matrix with the same number of off-diagonals, or substitutions, in each row with the substitutions drawn from the matrix row with the replacement. The A+T content of the stationary vector from this resampled matrix was calculated, and the average and standard deviation determined for 1000 resamples. Substitutions within tetranucleotide contexts from noncoding regions were taken from the previous analysis [
9]. All calculations were done using Python script written by the author.
4. Discussion
This study compared substitutions at fourfold degenerate (FFD) coding sites and noncoding (NC) intergenic sites, controlling for the composition of the tetranucleotide surrounding each substitution, or context-dependent effects [
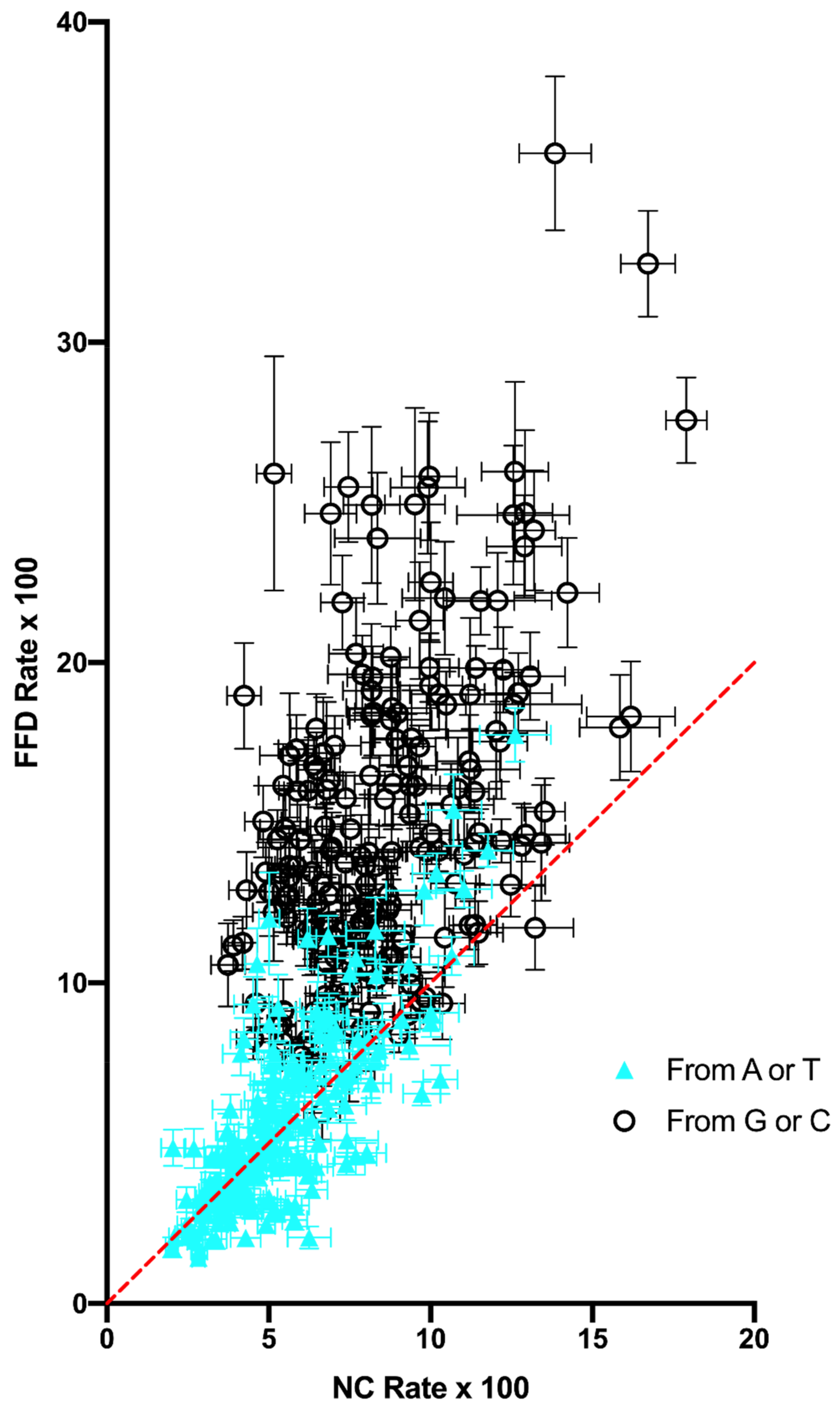
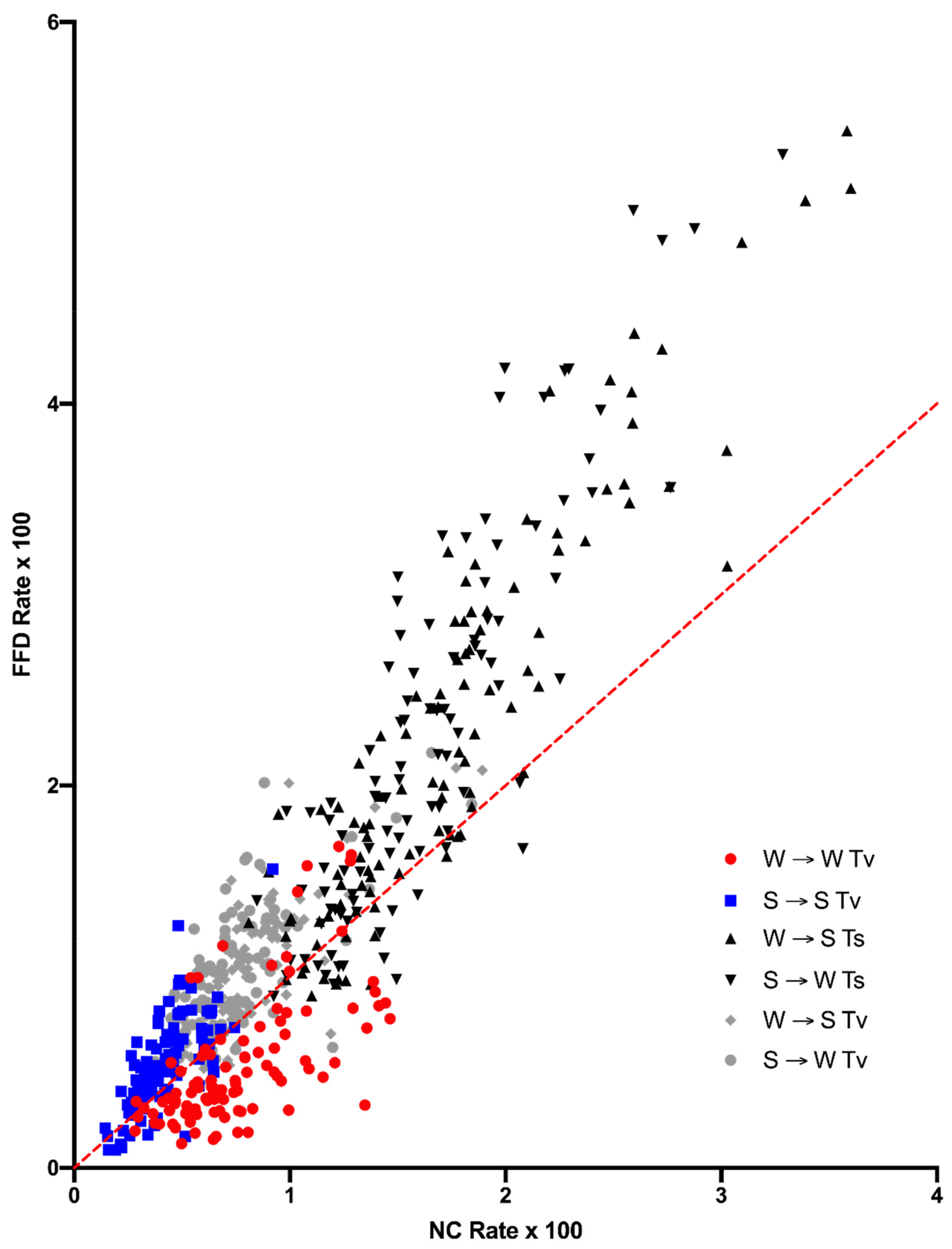
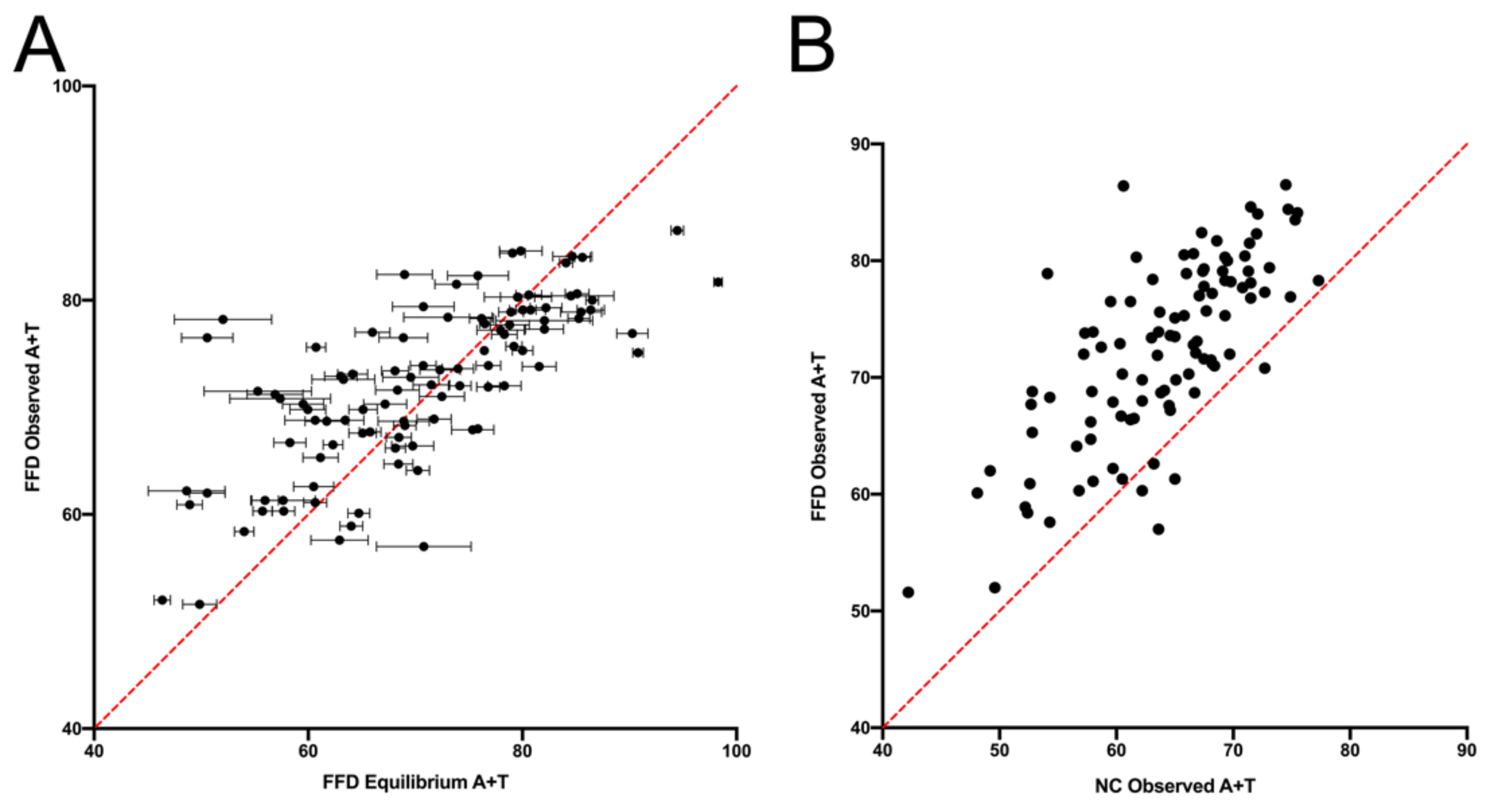
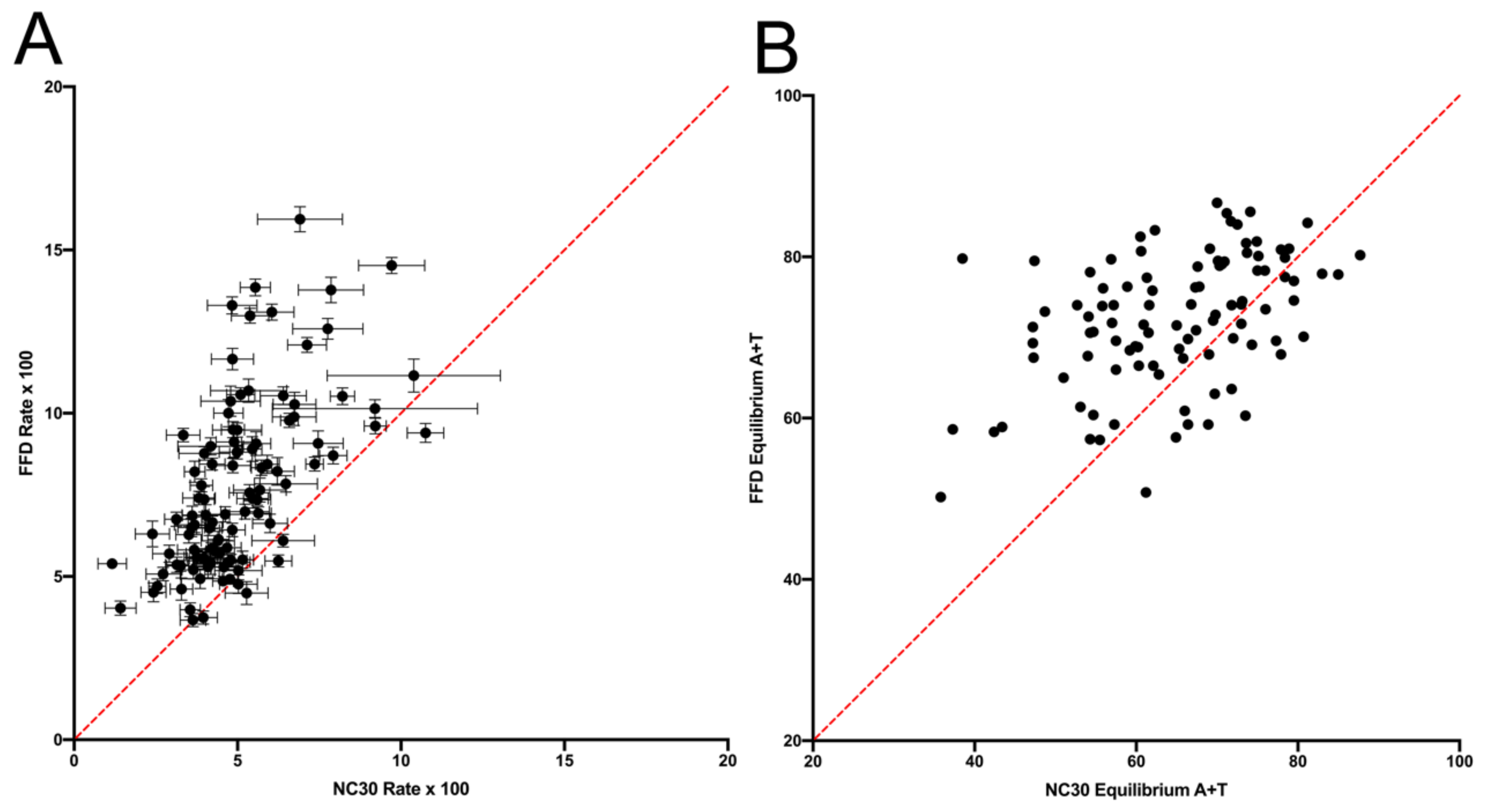
9]. The expectation is that, if mutation dynamics are the same at FFD and NC sites, then they should display the same context-dependent substitution dynamics in the absence of selection. It is shown that, although variation across sites is strongly correlated between the FFD and NC data, the results strongly suggest that there is some difference in substitution dynamics between them. The data show both that FFD sites have a higher rate of substitution, with the exception of W → W transversions, which have a higher rate at NC sites, and that the two types of sites differ in their substitution dynamics in some manner such that FFD sites are expected to evolve to a higher equilibrium A+T content, a prediction that is supported by compositional data. These two observations, which come from different ways of comparing the substitution data, are likely to be related in some way.
The most obvious possible explanation for the observed differences in substitution dynamics is that there are selective differences between NC and FFD. However, it is difficult to develop a selection model that could explain the data. There could be selective constraints on FFD sites, such as codon adaptation [
3,
20], effects of codon usage on protein folding [
21], and/or mRNA secondary structure [
22], but these would be expected to decrease the substitution rate, not increase it, relative to NC sites. The higher general rate of G and C substitutions at FFD sites could indicate positive selection for A and T or, conversely, selection against these bases at NC sites. However, the fact that NC sites have a lower rate of T → A and A → T transversions (
Figure 2) is not consistent with selection is simply for A+T content, nor is the difference in W → S and S → W transversions between the two site types. There is no obvious selection model to account for the increased rate of substitution at FFD sites with the sole exception of W → W transversions, which are actually decreased relative to NC sites.
A second difficulty with a selective explanation is that codon adaptation in flowering plant chloroplast genes is apparent in the atypical codon usage of the highly expressed
psbA gene, but there is no evidence for widespread adaptation across genes, which shows a general bias towards A and T at the synonymous positions [
3,
23]. Thus, selection for higher rates of G and C substitution would need to be the result of something other than selection for translation efficiency, either another form of selection on codon usage or on some factor other than codon usage, such as mRNA stability and influences on protein folding. It has long been established that Angiosperm chloroplasts have a low effective population size [
24] such that cpDNA is influenced by drift to a much higher degree than the nucleus [
25]. So, the substitution data would require either strong positive selection on mutations, with the exception of W → W transversions in coding regions, or selection against everything except these transversions in NC regions. Such strong selection would run counter to the observations about codon usage in cpDNA [
23].
While a selective explanation cannot be excluded, there is no adequate model to explain the substitution data here. In the absence of a good selective explanation, we considered possible neutral explanations that would be based on some difference in mutation dynamics between FFD and NC sites. This would not need to be a direct difference that results from functionality of a site, and there is no model for how such a difference could exist, but instead could be an indirect result of some difference that arises because of the functional differences.
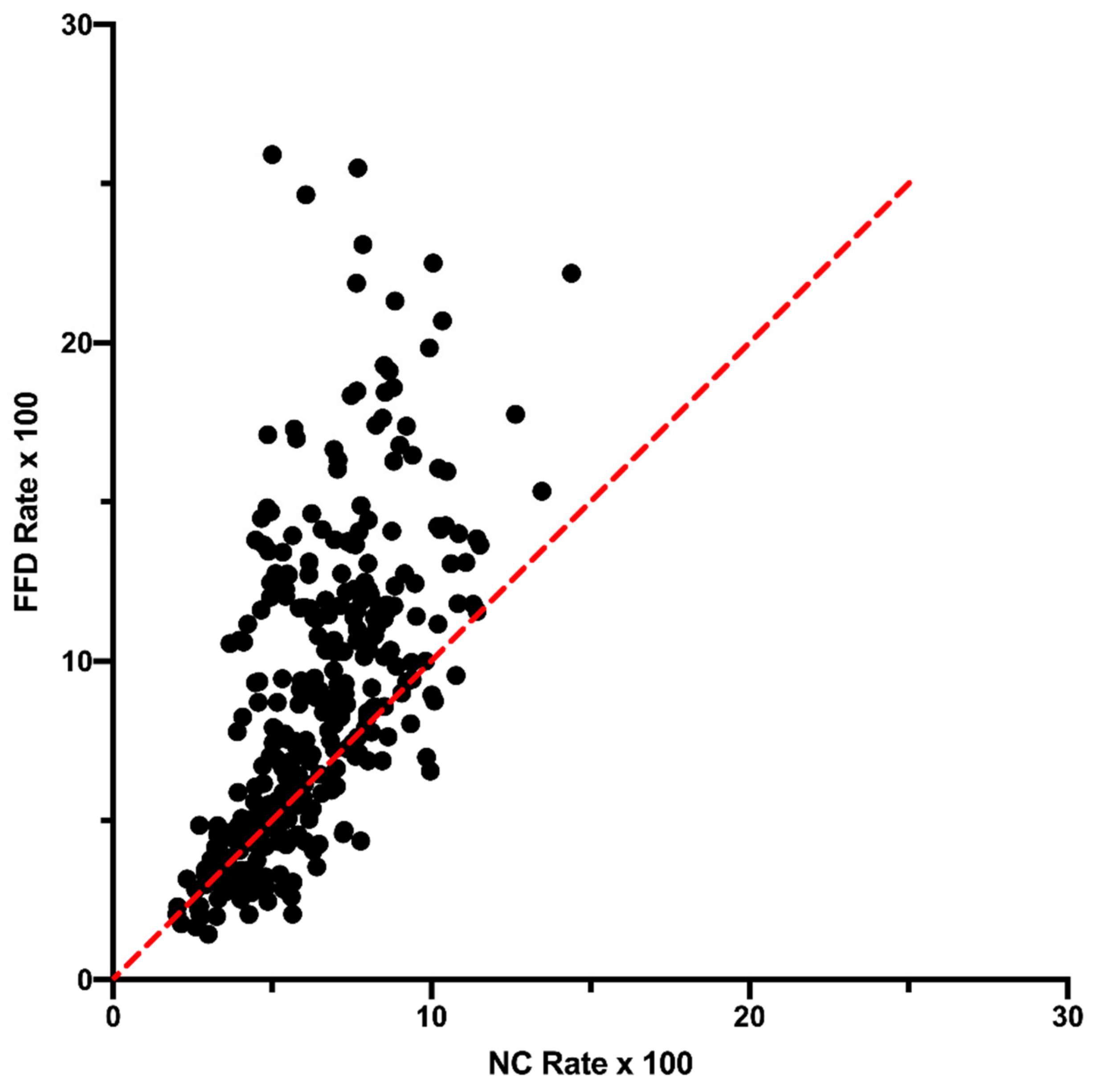
One neutral explanation could be that there is a difference in transcription level between the site types and this affects some aspect of mutation/repair [
13,
14]. This would be similar to the possibility that a difference in mutation and repair dynamics between leading and lagging strand contributes to strand asymmetry in some bacterial genomes [
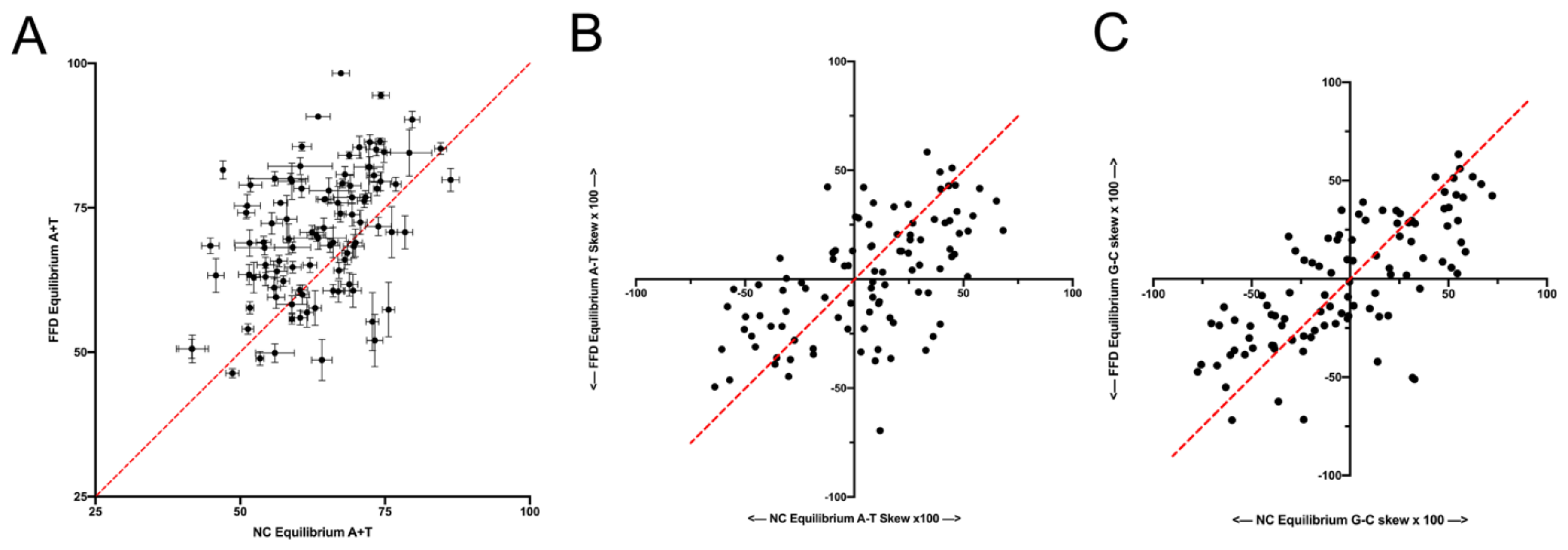
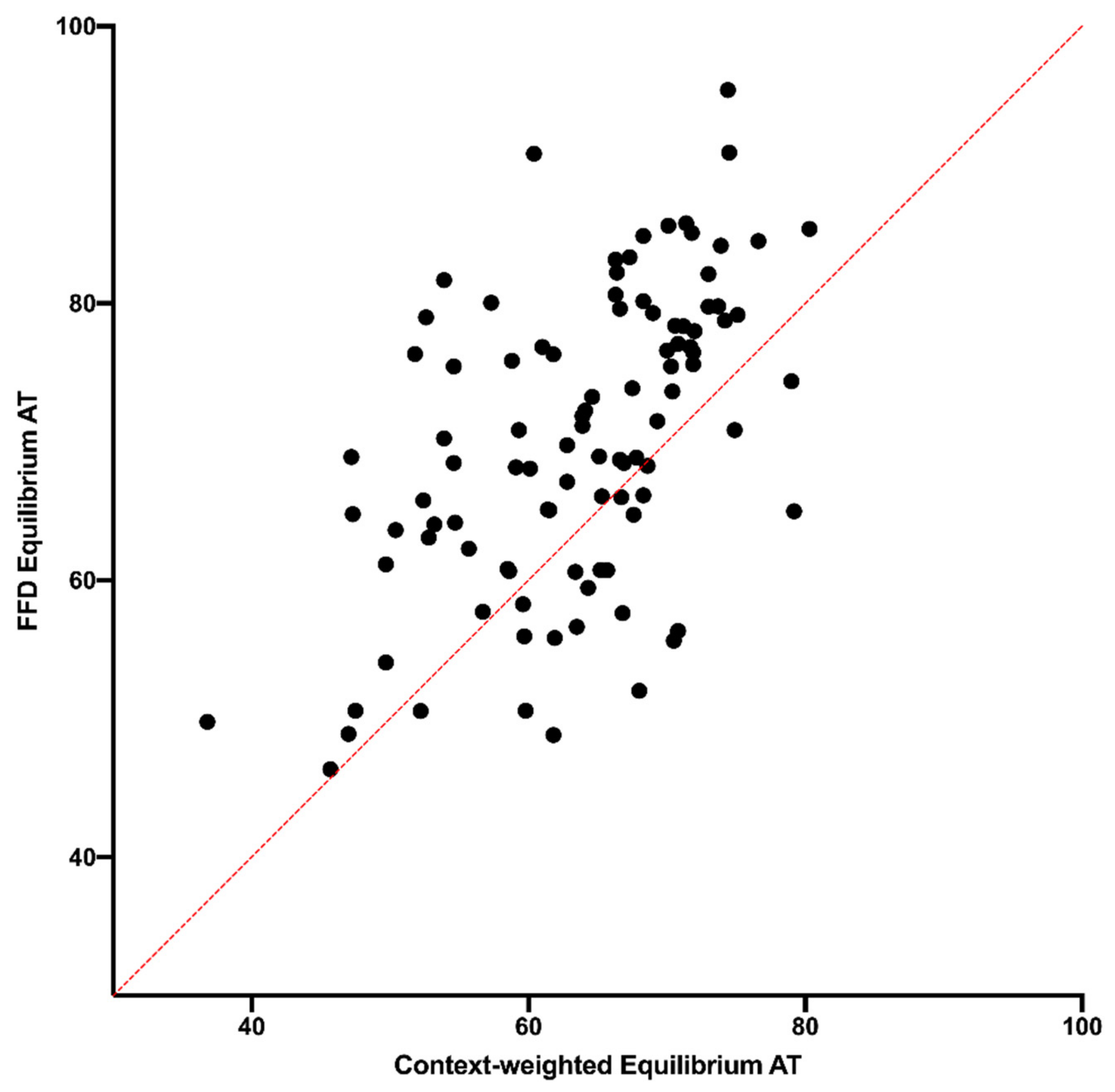
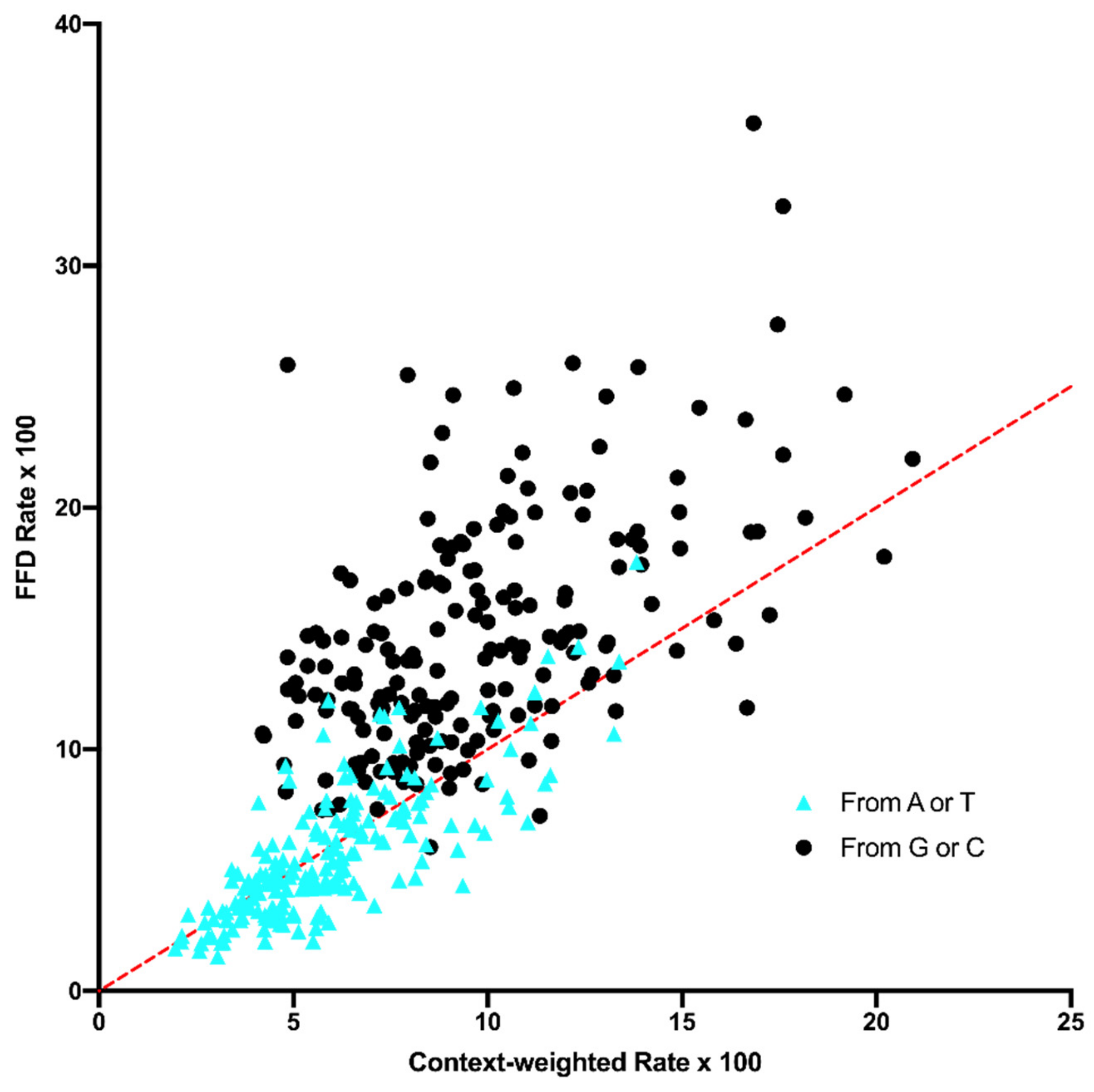
26]. The data from the NC30 analysis here show that, although that transcription might influence substitution dynamics to some degree, any effect cannot account for the difference between FFD and NC sites. However, a fuller examination of this possibility should be performed based on transcript mapping and levels. A second neutral explanation could be that the influence of context on mutation is more complex than what was controlled for. Even though the data indicate that the difference between FFD and NC sites exists when controlling for the composition of the surrounding hexanucleotide, there could be subtle effects of an even broader region, which, on average, differ in composition between coding and noncoding sites. The data in
Figure 6 and
Figure 7 suggest that wider context effects do not account for the difference between FFD and NC sites. This is perhaps not surprising since, as described in the Materials and Methods Section, the pentanucleotide {N
2 N
1 [N
0] N
1 N
2} is composed of the N
1 and N
2 neighboring base pairs that compose the tetranucleotide context flanking the site of substitution N
0. If we expand this to the heptanucleotide {N
3 N
2 N
1 [N
0] N
1 N
2 N
3}, where the N
0 substitution site is FFD, then the two bases that make up the N
3 neighboring pair would both be third codon position sites, which are generally A+T-rich like intergenic regions. Thus, the distribution of tetranucleotide contexts across hexanucleotide contexts are probably fairly similar for FFD and NC sites.
A third potential neutral explanation is that there are differences in epigenetic modification that give rise to differences in rates of specific types of mutation across sites, such as what we observe in the deamination of methylated CG sites [
15,
16,
17,
18]. The similarity in the increased rate of CT transitions in the CG context here (
Table 1) does not support any effect of CG methylation on the difference between FFD and NC sites. However, it is possible that there are other types of modification, such as CNG methylation, in cpDNA that differ between coding and noncoding DNA and which could give rise to a difference in underlying mutation bias. Particularly due to the difference between W → W and other types of substitution, this appears to be the most promising hypothesis to explain the substitution data in this study and should be explored in future analyses. It could also explain the rate differences between NC30 and NC sites (
Supplemental Figure S3) if these sites tend to differ in epigenetic modifications due to proximity to coding DNA.
Regardless of the underlying reason for the differences in substitution dynamics between FFD and NC sites, the observations here raise some caution for the use of either type of site as a proxy for neutral evolution. A comparison of exon and intron SNPs from the human genome suggested a similar phenomenon [
4], and although the results were not replicated using germline mutation data [
7], some profit should come from more thoroughly exploring the factors that influence mutation dynamics in various genomes. Of particular interest here, substitution data from NC sites have been used to explain codon usage bias in flowering plant genes, with the results indicating that selection is not necessary to explain codon usage except in the case of the
psbA gene [
10]. The basis of this was basically the fact that the composition of synonymous sites in coding genes was consistent with expected compositions generated using the context-dependent substitution patterns in intergenic regions. This is intriguing given the main findings here. Since the difference between FFD and NC sites is predominant in substitutions of G and C, and silent sites in chloroplast genes are strongly A+T-rich, it is quite possible that any difference between FFD and NC sites is too small to affect the use of NC substitutions to predict codon bias. However, further analysis is necessary to assess the use of intergenic regions to predict neutral composition patterns within coding sequences.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}