
Heterogeneous Genomic Divergence Landscape in Two Commercially Important European Scallop Species

Abstract
1. Introduction
2. Materials and Methods
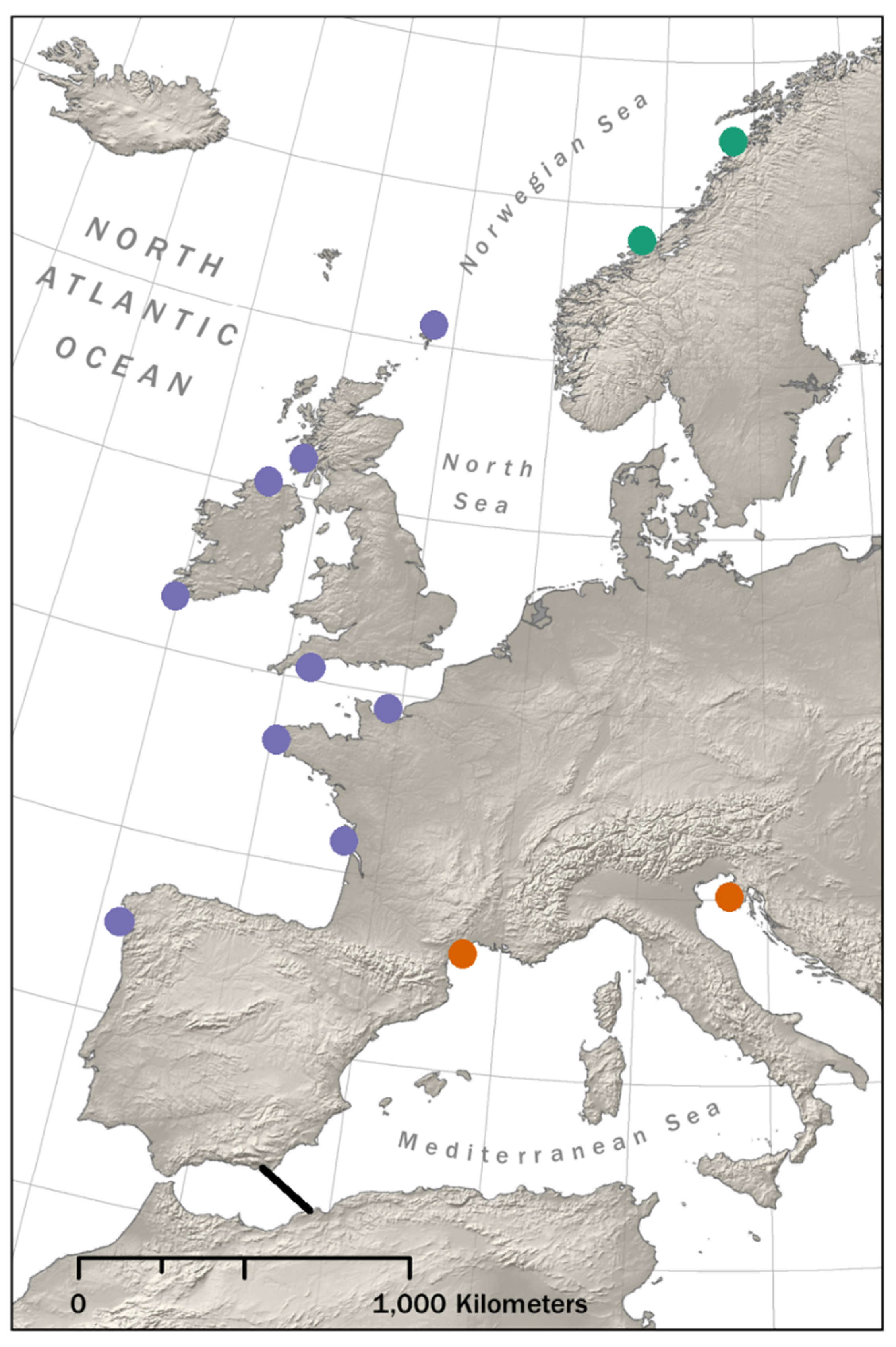
2.1. RAD Sequencing Data
2.2. Raw Read Processing, SNP Calling, and Genotyping
2.3. Demographic Modelling
2.4. Comparative Genomics
2.5. Linkage Disequilibrium
2.6. Gene Annotations and Enrichment Analysis
3. Results
3.1. Demographic Inference
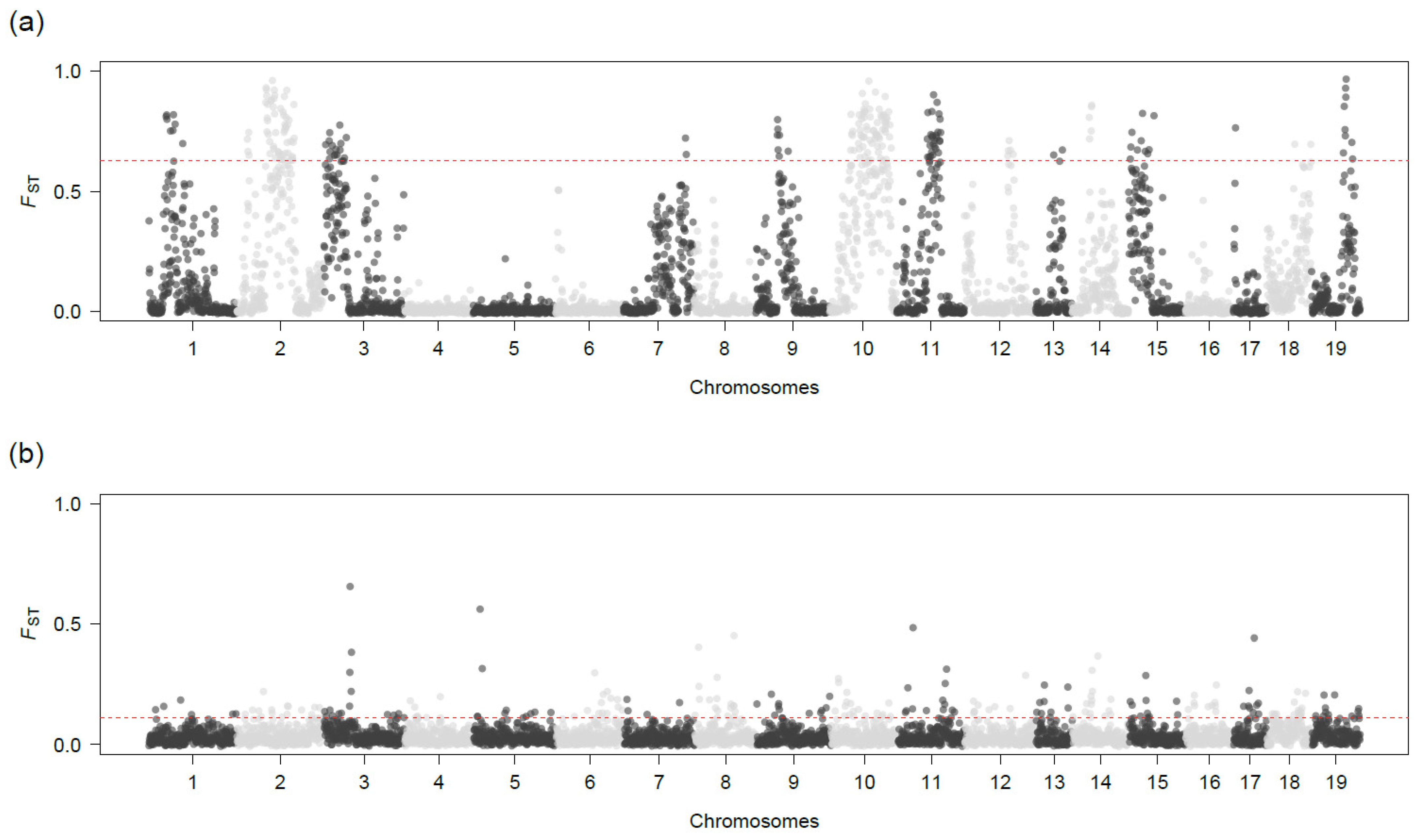
3.2. Genomic Divergence
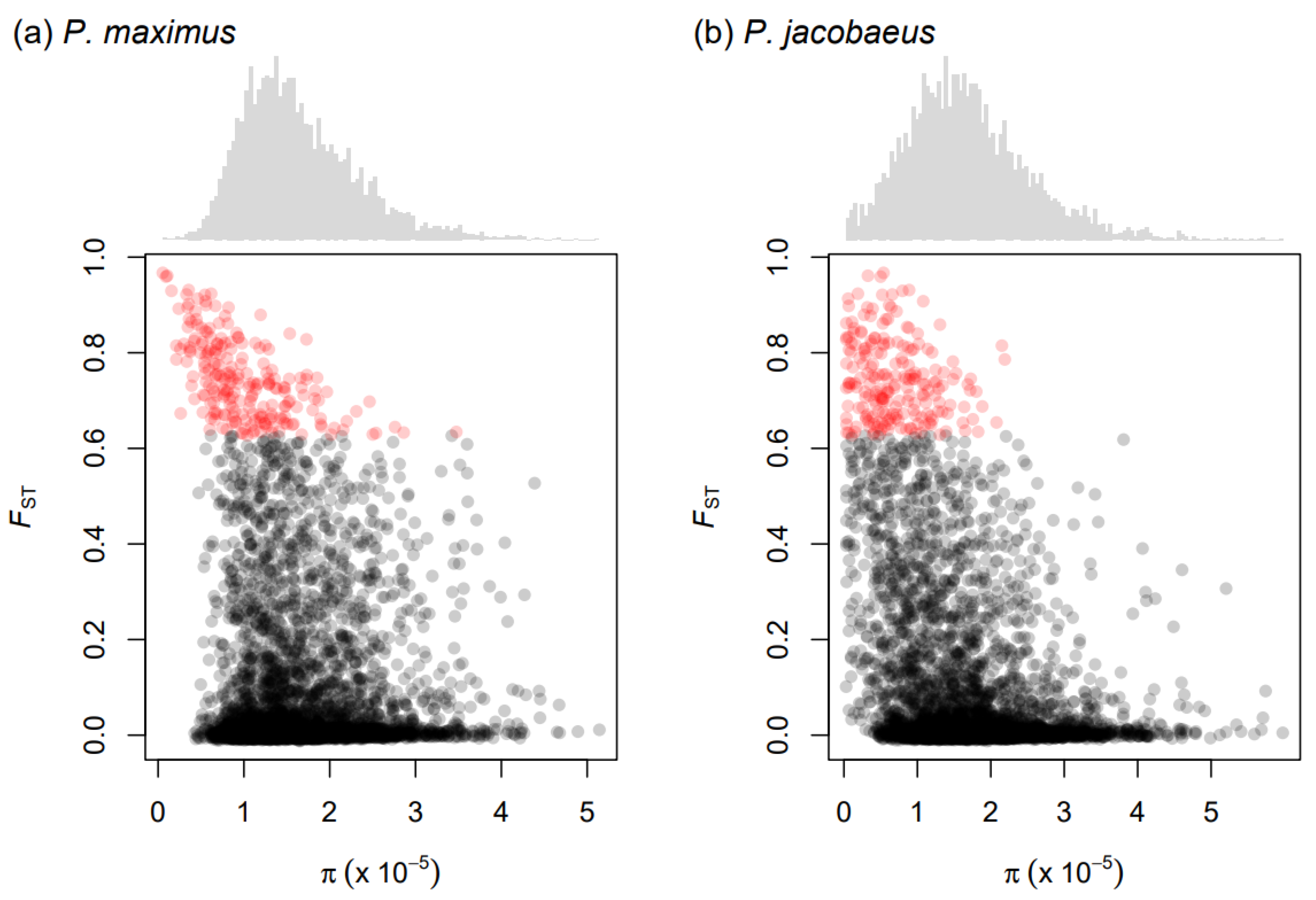
3.3. FST, Nucleotide Diversity, and dxy Values
3.4. Diagnostic Loci
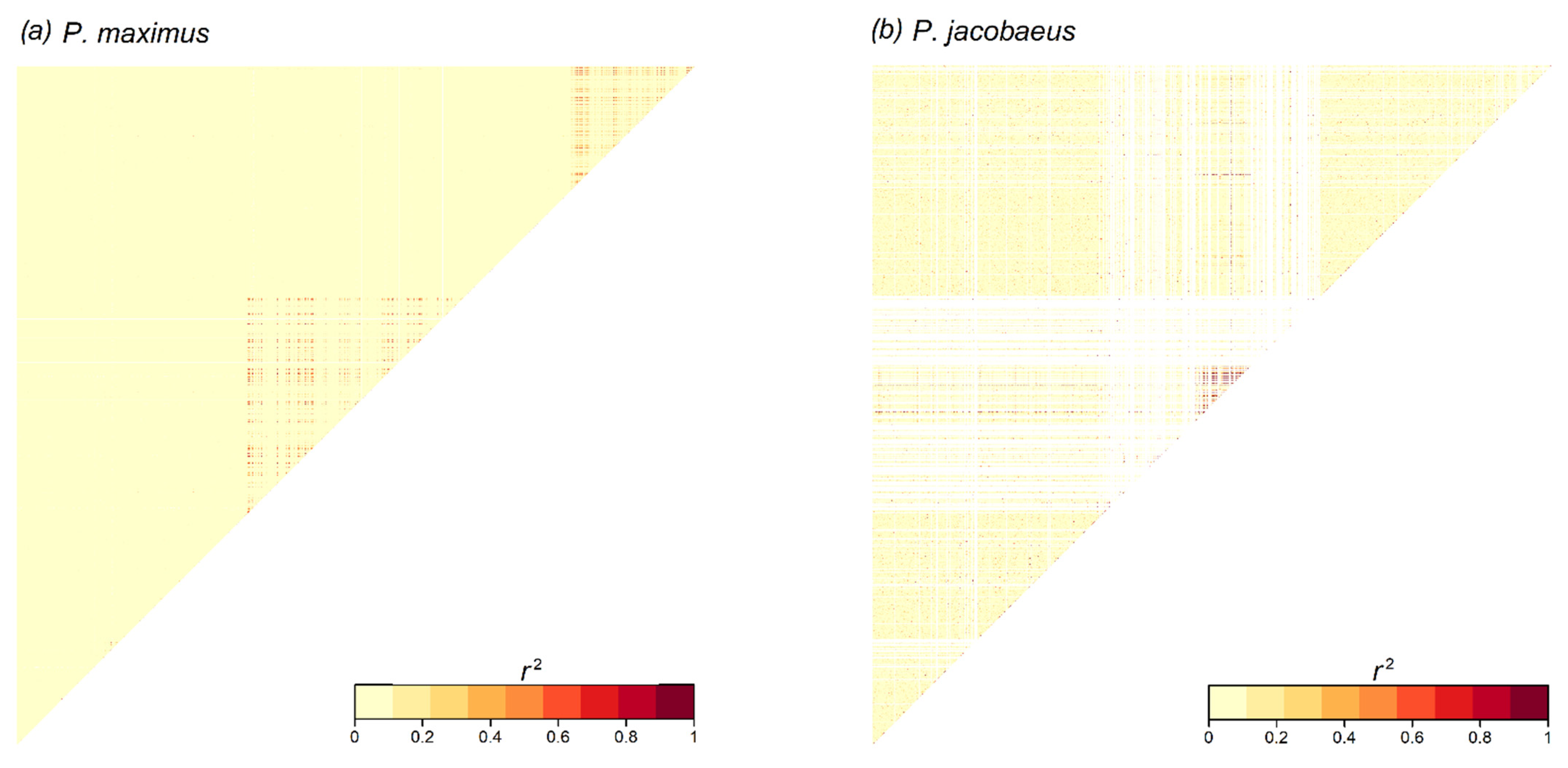
3.5. Linkage Disequilibrium
3.6. Gene Annotations and Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roux, C.; Fraïsse, C.; Romiguier, J.; Anciaux, Y.; Galtier, N.; Bierne, N. Shedding Light on the Grey Zone of Speciation along a Continuum of Genomic Divergence. PLoS Biol. 2016, 14, e2000234. [Google Scholar] [CrossRef]
- Seehausen, O.; Butlin, R.K.; Keller, I.; Wagner, C.E.; Boughman, J.W.; Hohenlohe, P.A.; Peichel, C.L.; Saetre, G.P.; Bank, C.; Brännström, Å.; et al. Genomics and the Origin of Species. Nat. Rev. Genet. 2014, 15, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Rundle, H.D.; Nosil, P. Ecological Speciation. Ecol. Lett. 2005, 8, 336–352. [Google Scholar] [CrossRef]
- Via, S.; West, J. The Genetic Mosaic Suggests a New Role for Hitchhiking in Ecological Speciation. Mol. Ecol. 2008, 17, 4334–4345. [Google Scholar] [CrossRef]
- Nielsen, R.; Williamson, S.; Kim, Y.; Hubisz, M.J.; Clark, A.G.; Bustamante, C. Genomic Scans for Selective Sweeps Using SNP Data. Genome Res. 2005, 15, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Noor, M.A.F.; Gratos, K.L.; Bertucci, L.A.; Reiland, J. Chromosomal Inversions and the Reproductive Isolation of Species. Proc. Natl. Acad. Sci. USA 2001, 98, 12084–12088. [Google Scholar] [CrossRef] [PubMed]
- Jones, F.C.; Grabherr, M.G.; Chan, Y.F.; Russell, P.; Mauceli, E.; Johnson, J.; Swofford, R.; Pirun, M.; Zody, M.C.; White, S.; et al. The Genomic Basis of Adaptive Evolution in Threespine Sticklebacks. Nature 2012, 484, 55–61. [Google Scholar] [CrossRef]
- Ravinet, M.; Faria, R.; Butlin, R.K.; Galindo, J.; Bierne, N.; Rafajlović, M.; Noor, M.A.F.; Mehlig, B.; Westram, A.M. Interpreting the Genomic Landscape of Speciation: A Road Map for Finding Barriers to Gene Flow. J. Evol. Biol. 2017, 30, 1450–1477. [Google Scholar] [CrossRef]
- Nosil, P.; Vines, T.H.; Funk, D.J. Perspective: Reproductive Isolation Caused by Natural Selection against Immigrants from Divergent Habitats. Evolution 2005, 59, 705–719. [Google Scholar] [CrossRef]
- Wright, S. Evolution in Mendelian Populations. Genetics 1931, 16, 97. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, T.E.; Hahn, M.W. Reanalysis Suggests That Genomic Islands of Speciation Are Due to Reduced Diversity, Not Reduced Gene Flow. Mol. Ecol. 2014, 23, 3133–3157. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Li, W.H. Mathematical Model for Studying Genetic Variation in Terms of Restriction Endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef]
- Ríos, C.; Sanz, S.; Saavedra, C.; Peña, J.B. Allozyme Variation in Populations of Scallops, Pecten jacobaeus (L.) and P. maximus (L.) (Bivalvia: Pectinidae), across the Almeria-Oran Front. J. Exp. Mar. Biol. Ecol. 2002, 267, 223–244. [Google Scholar] [CrossRef]
- Patarnello, T.; Volckaert, F.A.M.J.; Castilho, R. Pillars of Hercules: Is the Atlantic-Mediterranean Transition a Phylogeographical Break? Mol. Ecol. 2007, 16, 4426–4444. [Google Scholar] [CrossRef]
- Duran, S.; Palacín, C.; Becerro, M.A.; Turon, X.; Giribet, G. Genetic Diversity and Population Structure of the Commercially Harvested Sea Urchin Paracentrotus Lividus (Echinodermata, Echinoidea). Mol. Ecol. 2004, 13, 3317–3328. [Google Scholar] [CrossRef]
- Marie, A.D.; Lejeusne, C.; Karapatsiou, E.; Cuesta, J.A.; Drake, P.; Macpherson, E.; Bernatchez, L.; Rico, C. Implications for Management and Conservation of the Population Genetic Structure of the Wedge Clam Donax Trunculus across Two Biogeographic Boundaries. Sci. Rep. 2016, 6, 39152. [Google Scholar] [CrossRef]
- El Ayari, T.; Trigui El Menif, N.; Hamer, B.; Cahill, A.E.; Bierne, N. The Hidden Side of a Major Marine Biogeographic Boundary: A Wide Mosaic Hybrid Zone at the Atlantic–Mediterranean Divide Reveals the Complex Interaction between Natural and Genetic Barriers in Mussels. Heredity 2019, 122, 770–784. [Google Scholar] [CrossRef]
- El Ayari, T.; El Menif, N.T.; Saavedra, C.; Cordero, D.; Viard, F.; Bierne, N. Unexpected Mosaic Distribution of Two Hybridizing Sibling Lineages in the Teleplanically Dispersing Snail Stramonita Haemastoma Suggests Unusual Postglacial Redistribution or Cryptic Invasion. Ecol. Evol. 2017, 7, 9016–9026. [Google Scholar] [CrossRef]
- Naciri, M.; Lemaire, C.; Borsa, P.; Bonhomme, F. Genetic Study of the Atlantic/Mediterranean Transition in Sea Bass (Dicentrarchus labrax). J. Hered. 1999, 90, 591–596. [Google Scholar] [CrossRef]
- Wilding, C.S.; Beaumont, A.R.; Latchford, J.W. Are Pecten Maximus and Pecten jacobaeus Different Species? J. Mar. Biol. Assoc. U. K. 1999, 79, 949–952. [Google Scholar] [CrossRef]
- Morvezen, R.; Charrier, G.; Boudry, P.; Chauvaud, L.; Breton, F.; Strand, Ø.; Laroche, J. Genetic Structure of a Commercially Exploited Bivalve, the Great Scallop Pecten Maximus, along the European Coasts. Conserv. Genet. 2016, 17, 57–67. [Google Scholar] [CrossRef][Green Version]
- Vendrami, D.L.J.; De Noia, M.; Telesca, L.; Handal, W.; Charrier, G.; Boudry, P.; Eberhart-Phillips, L.; Hoffman, J.I. RAD Sequencing Sheds New Light on the Genetic Structure and Local Adaptation of European Scallops and Resolves Their Demographic Histories. Sci. Rep. 2019, 9, 7455. [Google Scholar] [CrossRef] [PubMed]
- Pérès, J.M. History of the Mediterranean Biota and the Colonization of the Depths. In Key Environments: Western Mediterranean; Pergamon Press: Oxford, UK, 1985; pp. 198–232. [Google Scholar]
- Raffi, S.; Stanley, S.M.; Marasti, R. Biogeographic Patterns and Plio-Pleistocene Extinction of Bivalvia in the Mediterranean and Southern North Sea. Paleobiology 1985, 11, 368–388. [Google Scholar] [CrossRef]
- Bateson, W. Heredity and Variation in Modern Lights. In Darwin and Modern Science; BoD—Books on Demand: Norderstedt, Germany, 1909. [Google Scholar]
- Dobzhansky, T.H. Studies on Hybrid Sterility. II. Localization of Sterility Factors in Drosophila Pseudoobscura Hybrids. Genetics 1936, 21, 113. [Google Scholar] [CrossRef]
- Muller, H. Isolating Mechanisms, Evolution and Temperature. Biol. Symp. 1942, 6, 71–125. [Google Scholar]
- Cutter, A.D. The Polymorphic Prelude to Bateson-Dobzhansky-Muller Incompatibilities. Trends Ecol. Evol. 2012, 27, 209–218. [Google Scholar] [CrossRef]
- Hollenbeck, C.M.; Portnoy, D.S.; Garcia, D.; Magnesen, T.; Matejusova, I.; Johnston, I.A.; Hollenbeck, C.M. Temperature-Associated Selection Linked to Putative Chromosomal Inversions in King Scallop (Pecten maximus). Proc. R. Soc. B 2022, 289, 20221573. [Google Scholar] [CrossRef]
- Kenny, N.J.; McCarthy, S.A.; Dudchenko, O.; James, K.; Betteridge, E.; Corton, C.; Dolucan, J.; Mead, D.; Oliver, K.; Omer, A.D.; et al. The Gene-Rich Genome of the Scallop Pecten Maximus. Gigascience 2020, 9, 1–13. [Google Scholar] [CrossRef]
- Zeng, Q.; Liu, J.; Wang, C.; Wang, H.; Zhang, L.; Hu, J.; Bao, L.; Wang, S. High-Quality Reannotation of the King Scallop Genome Reveals No ‘Gene-Rich’ Feature and Evolution of Toxin Resistance. Comput. Struct. Biotechnol. J. 2021, 19, 4954–4960. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual (No. Ed. 2); Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An Analysis Tool Set for Population Genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Excoffier, L.; Marchi, N.; Marques, D.A.; Matthey-Doret, R.; Gouy, A.; Sousa, V.C. Fastsimcoal2: Demographic Inference under Complex Evolutionary Scenarios. Bioinformatics 2021, 37, 4882–4885. [Google Scholar] [CrossRef]
- Varin, C.; Vidoni, P. A Note on Composite Likelihood Inference and Model Selection. Biometrika 2005, 92, 519–528. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; Bassham, S.; Etter, P.D.; Stiffler, N.; Johnson, E.A.; Cresko, W.A. Population Genomics of Parallel Adaptation in Threespine Stickleback Using Sequenced RAD Tags. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef]
- Korunes, K.L.; Samuk, K. Pixy: Unbiased Estimation of Nucleotide Diversity and Divergence in the Presence of Missing Data. Mol. Ecol. Resour. 2021, 21, 1359–1368. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Hill, W.G.; Weir, B.S. Variances and Covariances of Squared Linkage Disequilibria in Finite Populations. Theor. Popul. Biol. 1988, 33, 54–78. [Google Scholar] [CrossRef]
- Shin, J.H.; Blay, S.; McNeney, B.; Graham, J. LDheatmap: An R Function for Graphical Display of Pairwise Linkage Disequilibria between Single Nucleotide Polymorphisms. J. Stat. Softw. 2006, 16, 1–9. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. TopGO: Enrichment Analysis for Gene Ontology. R package version 2.48.0. 2022. [Google Scholar]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Saavedra, C.; Peña, J.B. Nucleotide Diversity and Pleistocene Population Expansion in Atlantic and Mediterranean Scallops (Pecten maximus and P. jacobaeus) as Revealed by the Mitochondrial 16S Ribosomal RNA Gene. J. Exp. Mar. Biol. Ecol. 2005, 323, 138–150. [Google Scholar] [CrossRef]
- Boon, E.; Faure, M.F.; Bierne, N. The Flow of Antimicrobial Peptide Genes through a Genetic Barrier between Mytilus edulis and M. galloprovincialis. J. Mol. Evol. 2009, 68, 461–474. [Google Scholar] [CrossRef]
- Tine, M.; Kuhl, H.; Gagnaire, P.A.; Louro, B.; Desmarais, E.; Martins, R.S.T.; Hecht, J.; Knaust, F.; Belkhir, K.; Klages, S.; et al. European Sea Bass Genome and Its Variation Provide Insights into Adaptation to Euryhalinity and Speciation. Nat. Commun. 2014, 5, 5770. [Google Scholar] [CrossRef]
- Le Moan, A.; Gagnaire, P.A.; Bonhomme, F. Parallel Genetic Divergence among Coastal-Marine Ecotype Pairs of European Anchovy Explained by Differential Introgression after Secondary Contact. Mol. Ecol. 2016, 25, 3187–3202. [Google Scholar] [CrossRef]
- Marth, G.T.; Czabarka, E.; Murvai, J.; Sherry, S.T. The Allele Frequency Spectrum in Genome-Wide Human Variation Three Large World Populations. Genetics 2004, 372, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Tintore, J.; La Violette, P.E.; Blade, I.; Cruzado, A. A Study of an Intense Density Front in the Eastern Alboran Sea: The Almeria–Oran Front. J. Phys. Oceanogr. 1988, 18, 1384–1397. [Google Scholar] [CrossRef]
- Duranton, M.; Allal, F.; Fraïsse, C.; Bierne, N.; Bonhomme, F.; Gagnaire, P.A. The Origin and Remolding of Genomic Islands of Differentiation in the European Sea Bass. Nat. Commun. 2018, 9, 2518. [Google Scholar] [CrossRef]
- Renaut, S.; Owens, G.L.; Rieseberg, L.H. Shared Selective Pressure and Local Genomic Landscape Lead to Repeatable Patterns of Genomic Divergence in Sunflowers. Mol. Ecol. 2014, 23, 311–324. [Google Scholar] [CrossRef]
- Ellegren, H.; Smeds, L.; Burri, R.; Olason, P.I.; Backström, N.; Kawakami, T.; Künstner, A.; Mäkinen, H.; Nadachowska-Brzyska, K.; Qvarnström, A.; et al. The Genomic Landscape of Species Divergence in Ficedula Flycatchers. Nature 2012, 491, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Berg, P.R.; Star, B.; Pampoulie, C.; Sodeland, M.; Barth, J.M.I.; Knutsen, H.; Jakobsen, K.S.; Jentoft, S. Three Chromosomal Rearrangements Promote Genomic Divergence between Migratory and Stationary Ecotypes of Atlantic Cod. Sci. Rep. 2016, 6, 23246. [Google Scholar] [CrossRef] [PubMed]
- Sodeland, M.; Jorde, P.E.; Lien, S.; Jentoft, S.; Berg, P.R.; Grove, H.; Kent, M.P.; Arnyasi, M.; Olsen, E.M.; Knutsen, H. “Islands of Divergence” in the Atlantic Cod Genome Represent Polymorphic Chromosomal Rearrangements. Genome Biol. Evol. 2016, 8, 1012–1022. [Google Scholar] [CrossRef]
- Faria, R.; Chaube, P.; Morales, H.E.; Larsson, T.; Lemmon, A.R.; Lemmon, E.M.; Rafajlović, M.; Panova, M.; Ravinet, M.; Johannesson, K.; et al. Multiple Chromosomal Rearrangements in a Hybrid Zone between Littorina Saxatilis Ecotypes. Mol. Ecol. 2019, 28, 1375–1393. [Google Scholar] [CrossRef] [PubMed]
- Wellenreuther, M.; Bernatchez, L. Eco-Evolutionary Genomics of Chromosomal Inversions. Trends Ecol. Evol. 2018, 33, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Cai, S.Y.; Shao, J.Z.; Chen, J. Toll-Like Receptors, Associated Biological Roles, and Signaling Networks in Non-Mammals. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Wang, M.; Yang, J.; Zhou, Z.; Ziu, L.; Wang, L.; Zhang, H.; Gao, Y.; Wang, X.; Zhang, L.; Zhao, J.; et al. A Primitive Toll-like Receptor Signaling Pathway in Mollusk Zhikong Scallop Chlamys Farreri. Dev. Comp. Immunol. 2011, 35, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Malmstrøm, M.; Matschiner, M.; Tørresen, O.K.; Star, B.; Snipen, L.G.; Hansen, T.F.; Baalsrud, H.T.; Nederbragt, A.J.; Hanel, R.; Salzburger, W.; et al. Evolution of the Immune System Influences Speciation Rates in Teleost Fishes. Nat. Genet. 2016, 48, 1204–1210. [Google Scholar] [CrossRef]
- Karvonen, A.; Seehausen, O. The Role of Parasitism in Adaptive Radiations-When Might Parasites Promote and When Might They Constrain Ecological Speciation? Int. J. Ecol. 2012, 2012. [Google Scholar] [CrossRef]
- Brennan, J.J.; Gilmore, T.D. Evolutionary Origins of Toll-like Receptor Signaling. Mol. Biol. Evol. 2018, 35, 1576–1587. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.; Guo, X.; Litman, G.W.; Dishaw, L.J.; Zhang, G. Massive Expansion and Functional Divergence of Innate Immune Genes in a Protostome. Sci. Rep. 2015, 5, 8693. [Google Scholar] [CrossRef]
- Leulier, F.; Lemaitre, B. Toll-like Receptors—Taking an Evolutionary Approach. Nat. Rev. Genet. 2008, 9, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, Writing and Erasing MRNA Methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.B. Gene Expression Drives Local Adaptation in Humans. Genome Res. 2013, 23, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Mendel, R.R. The Molybdenum Cofactor. J. Biol. Chem. 2013, 288, 13165–13172. [Google Scholar] [CrossRef]
- Pujolar, J.M.; Jacobsen, M.W.; Bertolini, F. Comparative Genomics and Signatures of Selection in North Atlantic Eels. Mar. Genom. 2022, 62, 100933. [Google Scholar] [CrossRef]
- Mack, K.L.; Nachman, M.W. Gene Regulation and Speciation. Trends Genet. 2017, 33, 68–80. [Google Scholar] [CrossRef]
- Goncalves, A.; Leigh-Brown, S.; Thybert, D.; Stefflova, K.; Turro, E.; Flicek, P.; Brazma, A.; Odom, D.T.; Marioni, J.C. Extensive Compensatory Cis-Trans Regulation in the Evolution of Mouse Gene Expression. Genome Res. 2012, 22, 2376–2384. [Google Scholar] [CrossRef]
- Davidson, J.H.; Balakrishnan, C.N. Gene Regulatory Evolution during Speciation in a Songbird. G3 Genes Genomes Genet. 2016, 6, 1357–1364. [Google Scholar] [CrossRef]
- McManus, C.J.; Coolon, J.D.; Duff, M.O.; Eipper-Mains, J.; Graveley, B.R.; Wittkopp, P.J. Regulatory Divergence in Drosophila Revealed by MRNA-Seq. Genome Res. 2010, 20, 816–825. [Google Scholar] [CrossRef]
- Tirosh, I.; Reikhav, S.; Levy, A.A.; Barkai, N. A Yeast Hybrid Provides Insight into the Evolution of Gene Expression Regulation. Science 2009, 324, 659–662. [Google Scholar] [CrossRef]
- Shi, X.; Ng, D.W.K.; Zhang, C.; Comai, L.; Ye, W.; Jeffrey Chen, Z. Cis- and Trans-Regulatory Divergence between Progenitor Species Determines Gene-Expression Novelty in Arabidopsis allopolyploids. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Huelvan, S. Variabilité Génétique de Populations de Pecten Maximus L. En Bretagne; Université de Bretagne Occidentale: Brest, France, 1985. [Google Scholar]
- Popović, N.T.; Krbavčić, M.; Barišić, J.; Ljubić, B.B.; Strunjak-Perović, I.; Babić, S.; Lorencin, V.; Matulić, D.; Tomljanović, T.; Čož-Rakovac, R. Comparative Tissue Responses of Marine Mollusks on Seasonal Changes in the Northern Adriatic Sea. Appl. Sci. 2021, 11, 2874. [Google Scholar] [CrossRef]
- Mestre, S.; Peña, J.B.; Farí, A.; Uriarte, I. The Spawning Period and Gametogenic Cycle of Pecten jacobeus (L.). Iberus 1990, 9, 161–167. [Google Scholar]
- Le Pennec, M.; Paugam, A.; Le Pennec, G. The Pelagic Life of the Pectinid Pecten Maximus—A Review. ICES J. Mar. Sci. 2003, 60, 211–223. [Google Scholar] [CrossRef]
- Boutet, I.; Moraga, D.; Marinovic, L.; Obreque, J.; Chavez-Crooker, P. Characterization of Reproduction-Specific Genes in a Marine Bivalve Mollusc: Influence of Maturation Stage and Sex on MRNA Expression. Gene 2008, 407, 130–138. [Google Scholar] [CrossRef]
- Sigang, F.; Yihui, G.; Youhou, X. Comparative Transcriptome Analysis of the Ovary and Testis in Noble Scallop (Chlamys nobilis). Pak. J. Zool. 2021, 53, 251. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | N | Highly Divergent | % Highly Divergent | Mean FST | Mean dxy | Diagnostic SNPs |
|---|---|---|---|---|---|---|
| 1 | 323 | 8 | 2.48 | 0.107 | 0.0045 | 0 |
| 2 | 321 | 59 | 18.38 | 0.259 | 0.0058 | 4 |
| 3 | 296 | 15 | 5.07 | 0.164 | 0.0051 | 0 |
| 4 | 253 | 0 | 0 | 0.007 | 0.0043 | 0 |
| 5 | 303 | 0 | 0 | 0.006 | 0.0040 | 0 |
| 6 | 250 | 0 | 0 | 0.017 | 0.0046 | 0 |
| 7 | 259 | 2 | 0.77 | 0.121 | 0.0046 | 0 |
| 8 | 228 | 0 | 0 | 0.031 | 0.0045 | 0 |
| 9 | 272 | 7 | 2.57 | 0.1 | 0.0045 | 0 |
| 10 | 246 | 70 | 28.46 | 0.398 | 0.0070 | 2 |
| 11 | 250 | 28 | 11.2 | 0.164 | 0.0054 | 54 |
| 12 | 262 | 5 | 1.91 | 0.079 | 0.0046 | 0 |
| 13 | 133 | 2 | 1.5 | 0.087 | 0.0046 | 0 |
| 14 | 209 | 5 | 2.39 | 0.108 | 0.0048 | 1 |
| 15 | 209 | 10 | 4.78 | 0.168 | 0.0058 | 0 |
| 16 | 176 | 0 | 0 | 0.02 | 0.0049 | 0 |
| 17 | 122 | 1 | 0.82 | 0.034 | 0.0047 | 0 |
| 18 | 166 | 2 | 1.21 | 0.169 | 0.0057 | 1 |
| 19 | 179 | 9 | 5.03 | 0.127 | 0.0057 | 0 |
| Unplaced scaffolds | 3 | 0 | 0 | 0.016 | 0.0049 | 0 |
| Total | 4460 | 223 | 5.0 | 0.118 | 0.0050 | 62 |
| GO Term ID | Description | p-Value |
|---|---|---|
| GO:0002755 | MyD88-dependent toll-like receptor signalling pathway | <0.01 |
| GO:0009058 | biosynthetic process | <0.01 |
| GO:0034472 | snRNA 3’-end processing | <0.05 |
| GO:0032456 | endocytic recycling | <0.05 |
| GO:0036265 | RNA (guanine-N7)-methylation | <0.05 |
| GO:0006777 | Mo-molybdopterin cofactor biosynthetic process | <0.05 |
| GO:0006465 | signal peptide processing | <0.05 |
| SNP Type | Within Gene | Transcribed | Highly Divergent | Diagnostic |
|---|---|---|---|---|
| intergenic region | N | N | 724 (0.41) | 24 (0.39) |
| downstream gene variant | N | N | 166 (0.09) | 10 (0.16) |
| upstream gene variant | N | N | 196 (0.11) | 5 (0.08) |
| 3’ UTR variant | Y | N | 34 (0.02) | 4 (0.06) |
| 5’ UTR variant | Y | N | 6 (0.003) | 0 (0) |
| intron variant | Y | N | 531 (0.3) | 18 (0.29) |
| splice region variant and intron variant | Y | N | 11 (0.01) | 0 (0) |
| synonymous variant | Y | Y | 67 (0.04) | 1 (0.02) |
| missense variant | Y | Y | 40 (0.02) | 0 (0) |
| loss of function | Y | Y | 2 (0.001) | 0 (0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vendrami, D.L.J.; Hoffman, J.I.; Wilding, C.S. Heterogeneous Genomic Divergence Landscape in Two Commercially Important European Scallop Species. Genes 2023, 14, 14. https://doi.org/10.3390/genes14010014
Vendrami DLJ, Hoffman JI, Wilding CS. Heterogeneous Genomic Divergence Landscape in Two Commercially Important European Scallop Species. Genes. 2023; 14(1):14. https://doi.org/10.3390/genes14010014
Chicago/Turabian StyleVendrami, David L. J., Joseph I. Hoffman, and Craig S. Wilding. 2023. "Heterogeneous Genomic Divergence Landscape in Two Commercially Important European Scallop Species" Genes 14, no. 1: 14. https://doi.org/10.3390/genes14010014
APA StyleVendrami, D. L. J., Hoffman, J. I., & Wilding, C. S. (2023). Heterogeneous Genomic Divergence Landscape in Two Commercially Important European Scallop Species. Genes, 14(1), 14. https://doi.org/10.3390/genes14010014