

Simultaneous SARS-CoV-2 Genome Sequencing of 384 Samples on an Illumina MiSeq Instrument through Protocol Optimization

 , , , and
, , , and 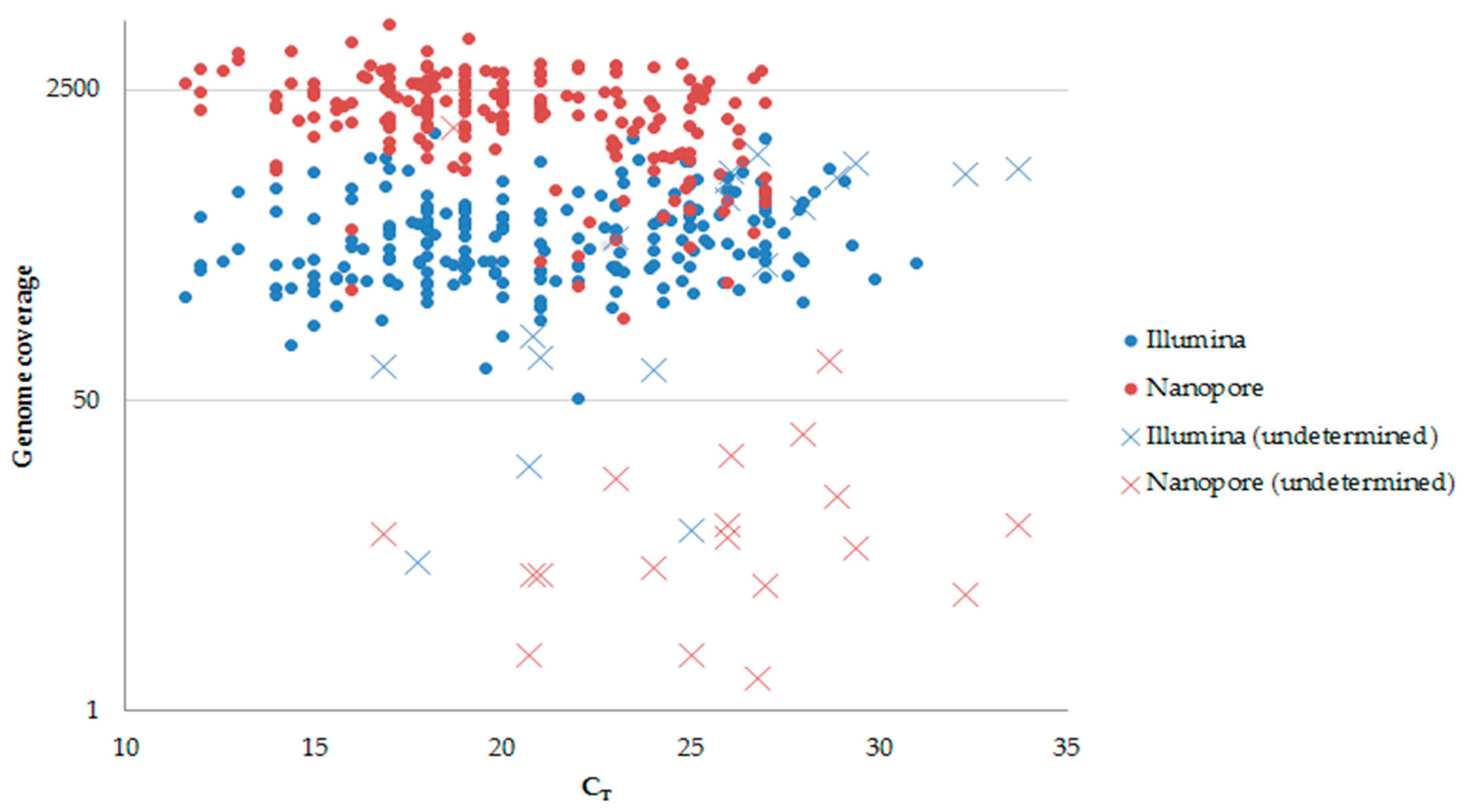{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. RNA Extraction
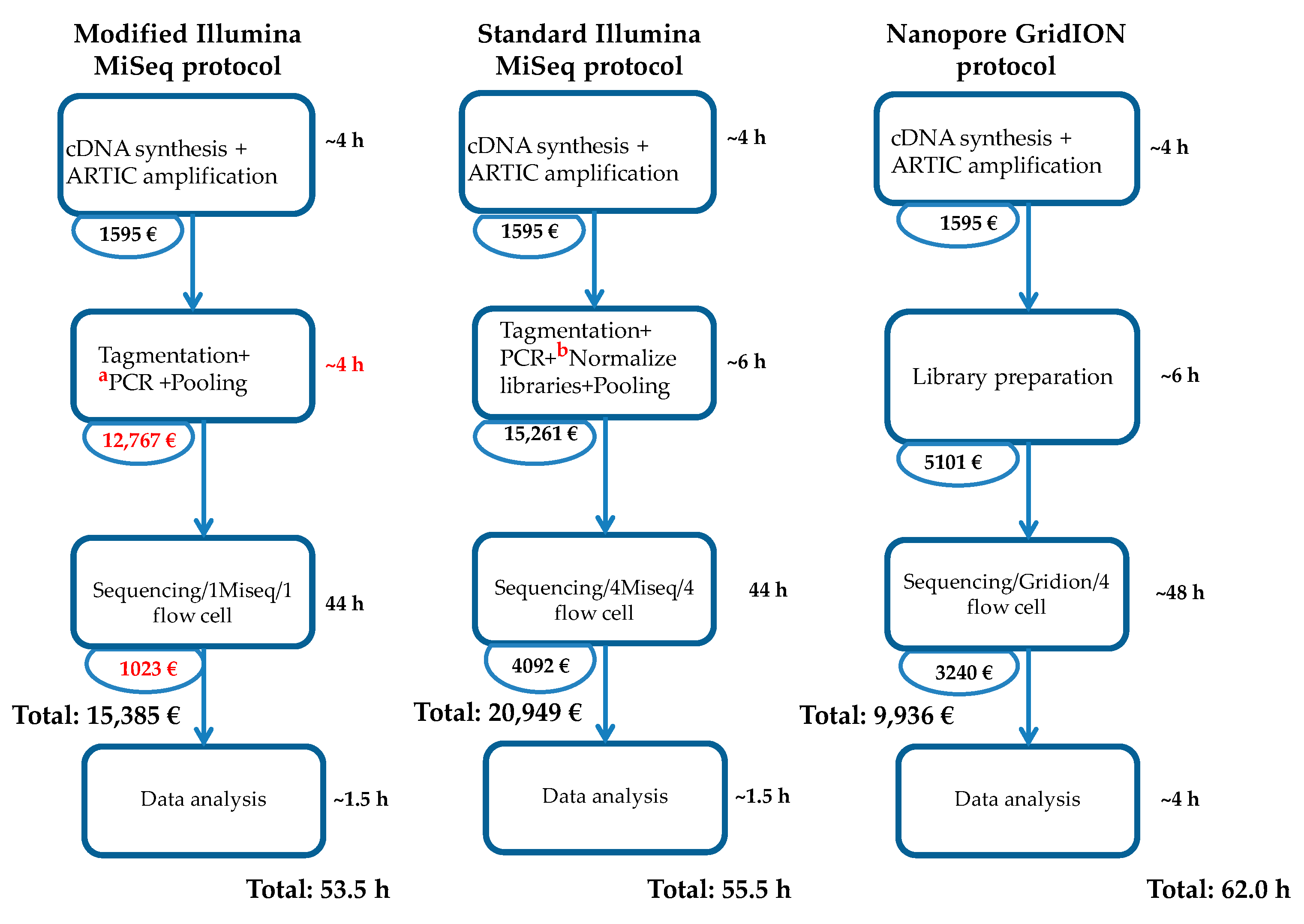
2.3. Modified Illumina MiSeq Protocol
2.3.1. cDNA Synthesis
2.3.2. Amplification of cDNA
2.3.3. Tagmentation of Genomic DNA
2.3.4. Amplification, Clean up and Pooling of Libraries
2.3.5. Sequencing of the Libraries
2.4. Nanopore Protocol
2.5. Genome Assembly, Variant Calling, and Phylogenetic Analysis
2.6. Illumina Sequencing Data Analysis
2.7. Nanopore Sequencing Data Analysis
2.8. Consensus Sequences Analysis
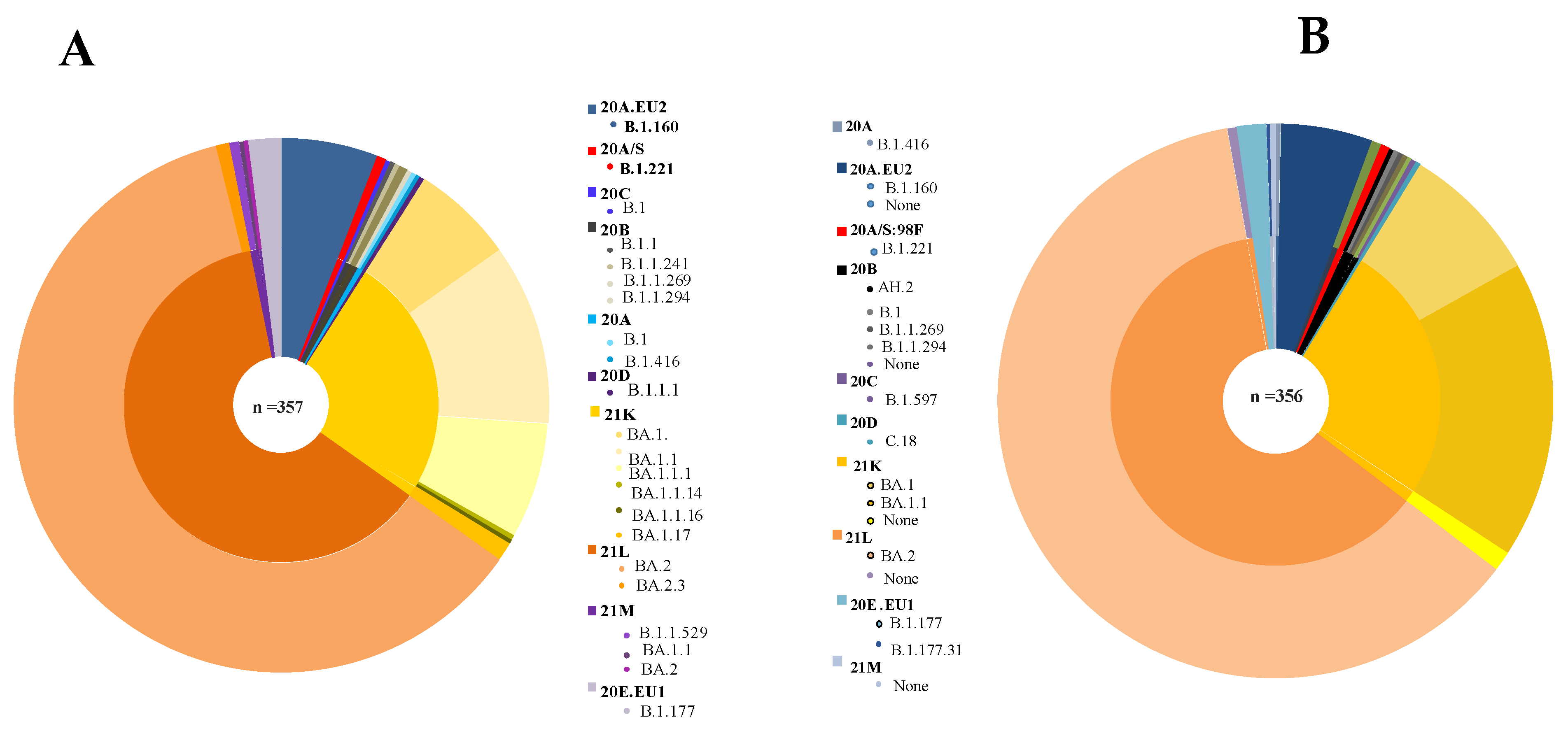
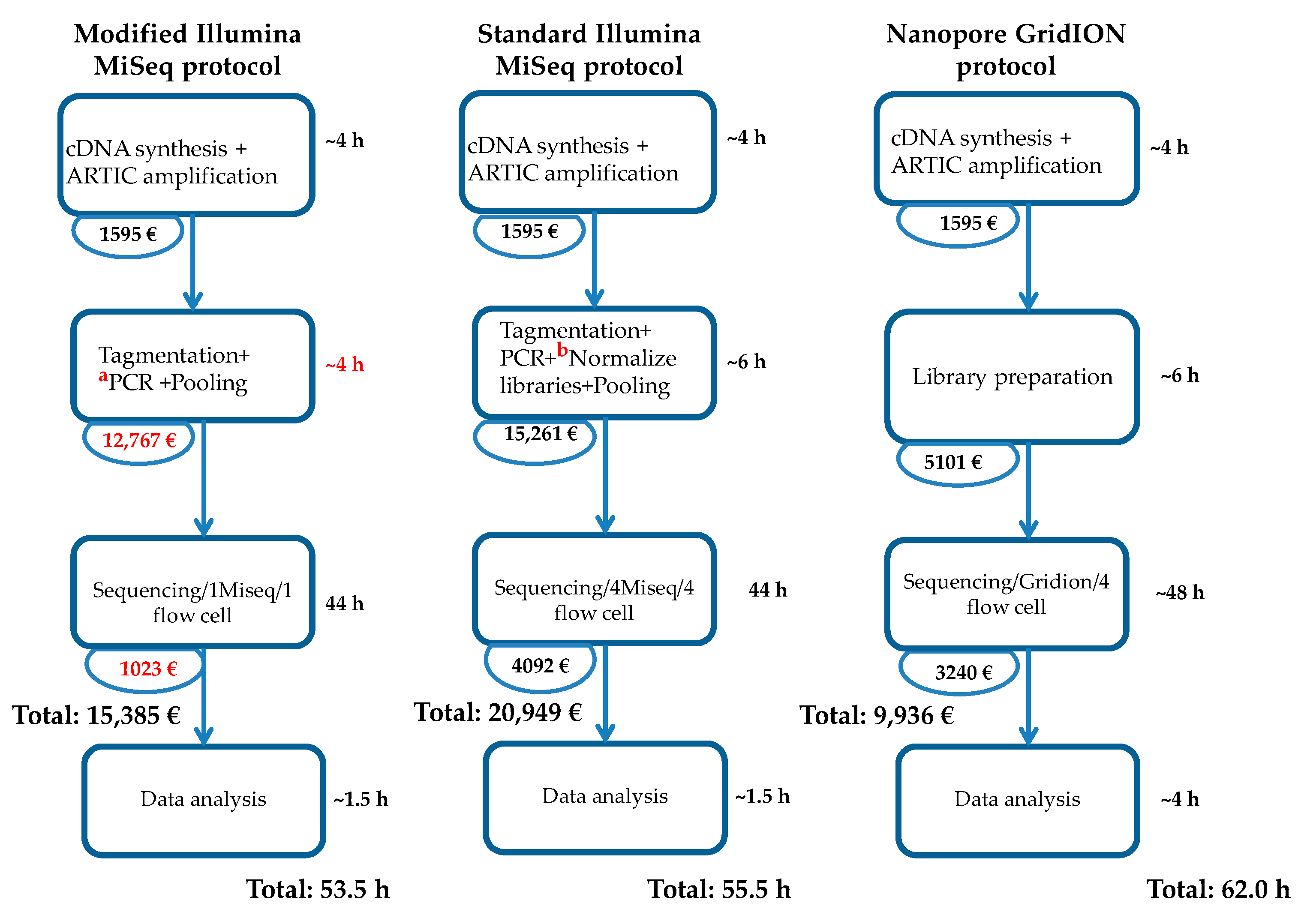
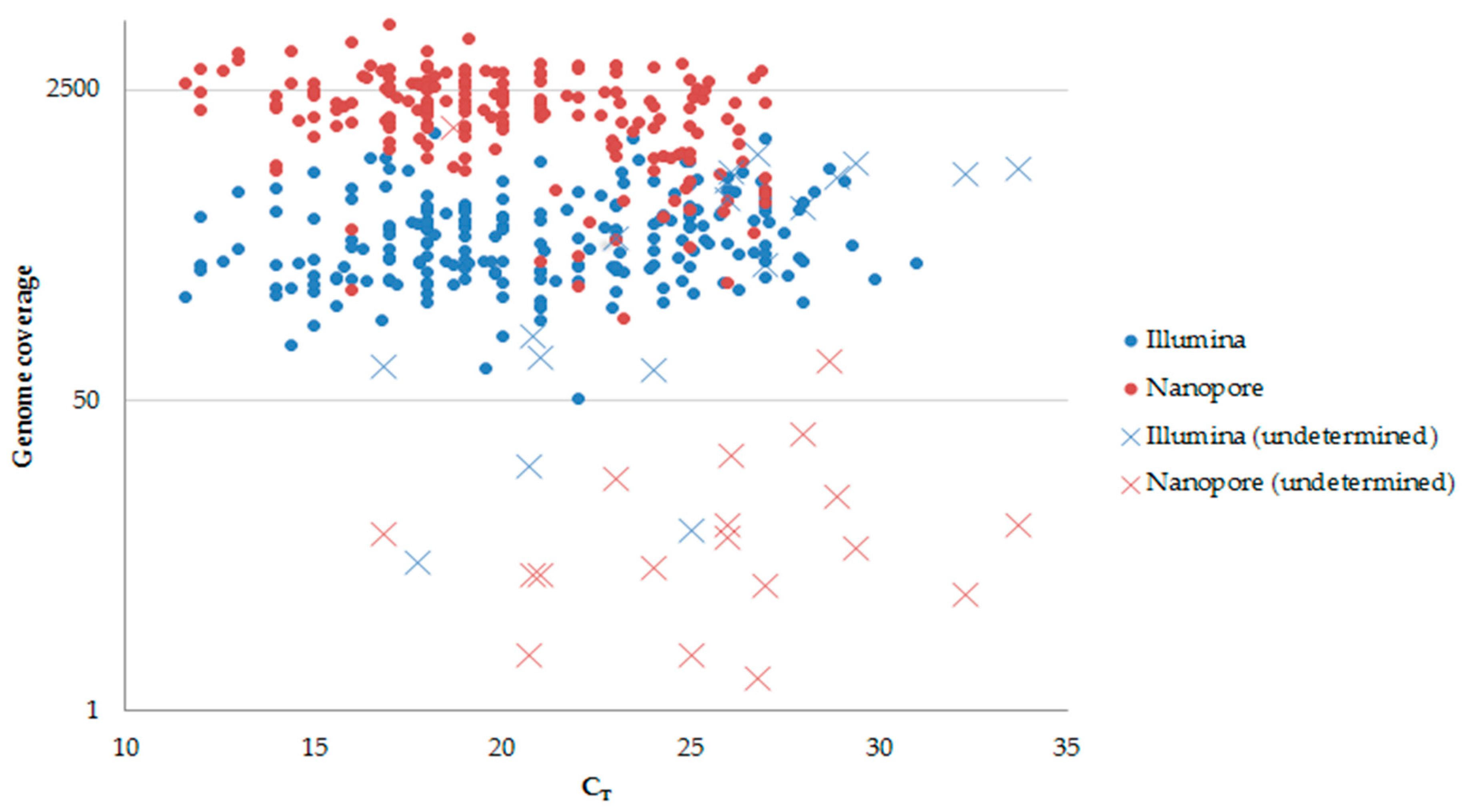
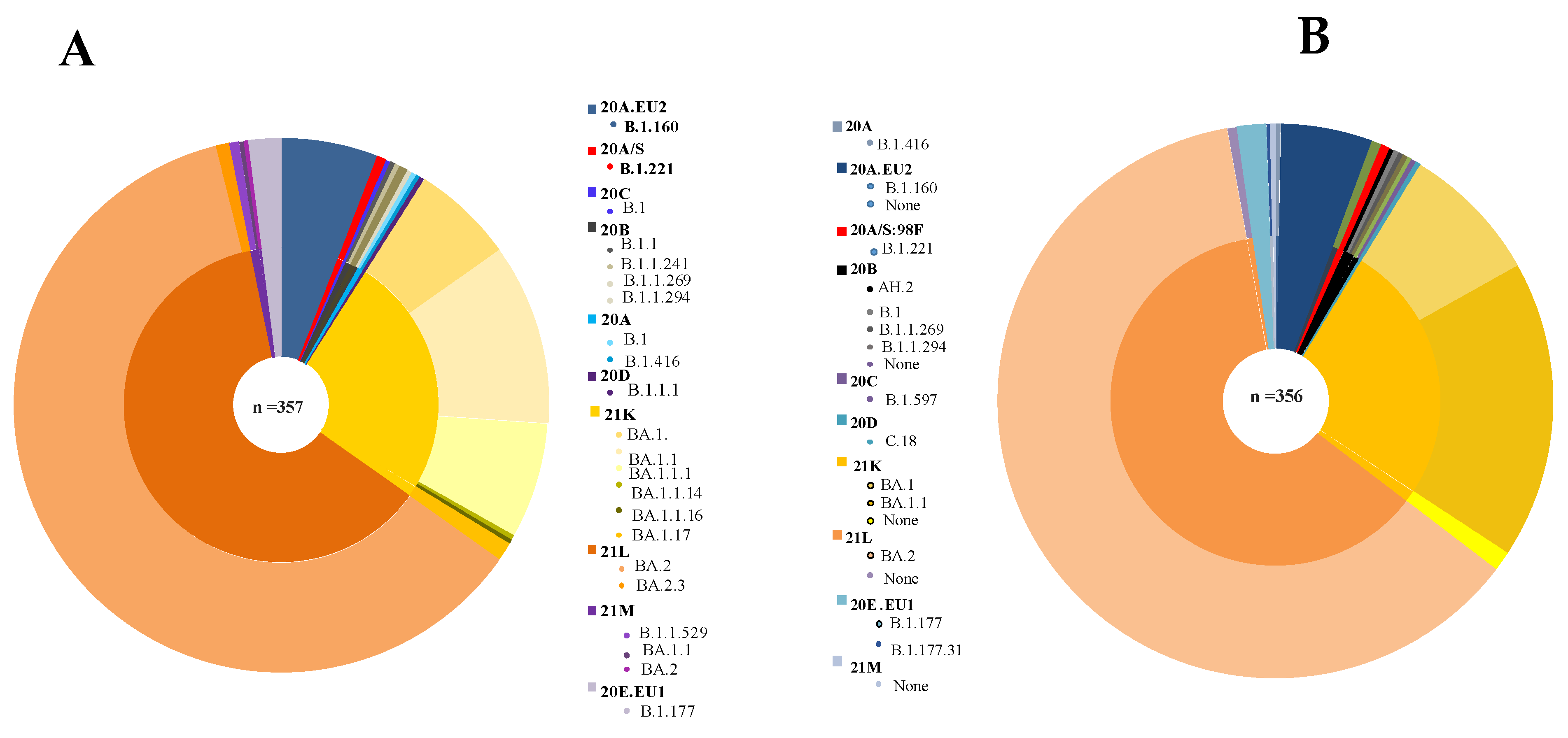
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Estimates of Excess Mortality Associated with COVID-19 Pandemic (as of 25 March 2022); WHO: Geneva, Switzerland, 2022. [Google Scholar]
- Charre, C.; Ginevra, C.; Sabatier, M.; Regue, H.; Destras, G.; Brun, S.; Burfin, G.; Scholtes, C.; Morfin, F.; Valette, M.; et al. Evaluation of NGS-based approaches for SARS-CoV-2 whole genome characterisation. Virus Evol. 2020, 6, veaa075. [Google Scholar] [CrossRef] [PubMed]
- ARTIC Network. Available online: https://artic.network (accessed on 11 May 2022).
- ARTIC Network. Available online: https://github.com/artic-network/artic-ncov2019/tree/master/primer_schemes/nCoV-2019/V4.1 (accessed on 13 September 2022).
- Franco-Munoz, C.; Alvarez-Diaz, D.A.; Laiton-Donato, K.; Wiesner, M.; Escandon, P.; Usme-Ciro, J.A.; Franco-Sierra, N.D.; Flórez-Sánchez, A.C.; Sergio Gómez-Rangel, S.; Rodríguez-Calderon, L.D.; et al. Substitutions in Spike and Nucleocapsid proteins of SARS-CoV-2 circulating in South America. Infect. Genet. Evol. 2020, 85, 104557. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv 2020. [Google Scholar]
- Bhoyar, R.C.; Senthivel, V.; Jolly, B.; Imran, M.; Jain, A.; Divakar, M.K.; Scaria, V.; Sivasubbu, S. An optimized, amplicon-based approach for sequencing of SARS-CoV-2 from patient samples using COVIDSeq assay on Illumina MiSeq sequencing platforms. STAR Protoc. 2021, 2, 100755. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://nanoporetech.com/resource-centre/overview-eco-pcr-tiling-sars-cov-2-virus (accessed on 1 June 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Available online: https://github.com/artic-network/artic-ncov2019 (accessed on 1 June 2022).
- ARTIC SARS-CoV-2 Bioinformatics Protocol. Available online: https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html (accessed on 1 June 2022).
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source 2021, 6, 3773. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Ã.H.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Bhoyar, R.C.; Jain, A.; Sehgal, P.; Divakar, M.K.; Sharma, D.; Imran, M.; Jolly, B.; Ranjan, G.; Rophina, M.; Sharma, S.; et al. High throughput detection and genetic epidemiology of SARS-CoV-2 using COVIDSeq next-generation sequencing. PLoS ONE 2021, 16, 2. [Google Scholar]
- Pillay, S.; Giandhari, J.; Tegally, H.; Wilkinson, E.; Chimukangara, B.; Lessells, R.; Moosa, Y.; Mattison, S.; Gazy, I.; Fish, M.; et al. Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic. Genes 2020, 11, 949. [Google Scholar] [CrossRef] [PubMed]
- WHO. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/fr/activities/tracking-SARS-CoV-2-variants (accessed on 1 May 2022).
- Plitnick, J.; Griesemer, S.; Lasek-Nesselquist, E.; Singh, N.; Lamson, D.M.; George, K.S. Whole-Genome Sequencing of SARS-CoV-2: Assessment of the Ion Torrent AmpliSeq Panel and Comparison with the Illumina MiSeq ARTIC Protocol. J. Clin. Microbiol. 2021, 59, 12. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papa Mze, N.; Beye, M.; Kacel, I.; Tola, R.; Basco, L.; Bogreau, H.; Colson, P.; Fournier, P.-E. Simultaneous SARS-CoV-2 Genome Sequencing of 384 Samples on an Illumina MiSeq Instrument through Protocol Optimization. Genes 2022, 13, 1648. https://doi.org/10.3390/genes13091648
Papa Mze N, Beye M, Kacel I, Tola R, Basco L, Bogreau H, Colson P, Fournier P-E. Simultaneous SARS-CoV-2 Genome Sequencing of 384 Samples on an Illumina MiSeq Instrument through Protocol Optimization. Genes. 2022; 13(9):1648. https://doi.org/10.3390/genes13091648
Chicago/Turabian StylePapa Mze, Nasserdine, Mamadou Beye, Idir Kacel, Raphael Tola, Leonardo Basco, Hervé Bogreau, Philippe Colson, and Pierre-Edouard Fournier. 2022. "Simultaneous SARS-CoV-2 Genome Sequencing of 384 Samples on an Illumina MiSeq Instrument through Protocol Optimization" Genes 13, no. 9: 1648. https://doi.org/10.3390/genes13091648
APA StylePapa Mze, N., Beye, M., Kacel, I., Tola, R., Basco, L., Bogreau, H., Colson, P., & Fournier, P.-E. (2022). Simultaneous SARS-CoV-2 Genome Sequencing of 384 Samples on an Illumina MiSeq Instrument through Protocol Optimization. Genes, 13(9), 1648. https://doi.org/10.3390/genes13091648