
Loss of Protein Function Causing Severe Phenotypes of Female-Restricted Wieacker Wolff Syndrome due to a Novel Nonsense Mutation in the ZC4H2 Gene
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Compliance
2.2. Next-Generation Sequencing and Sanger Sequencing
2.3. X-Chromosome Inactivation (XCI) Assay
2.4. Domain and Secondary Structure Analysis of De Novo Nonsense Mutation
2.5. Construction of the ZC4H2 Expression Vectors
2.6. Cell Transfection and Immunoblot Analysis
2.7. Lentivirus Vectors for ZC4H2 Small Hairpin RNA (shRNA)
2.8. Induced Differentiation of Neural Stem Cells (NSCs) and Lentivirus shRNA Gene Transfection
2.9. RNA Sequencing (RNA-Seq) and Processing RNA Seq Data
2.10. PCR Array Analysis
2.11. RT-qPCR
3. Results
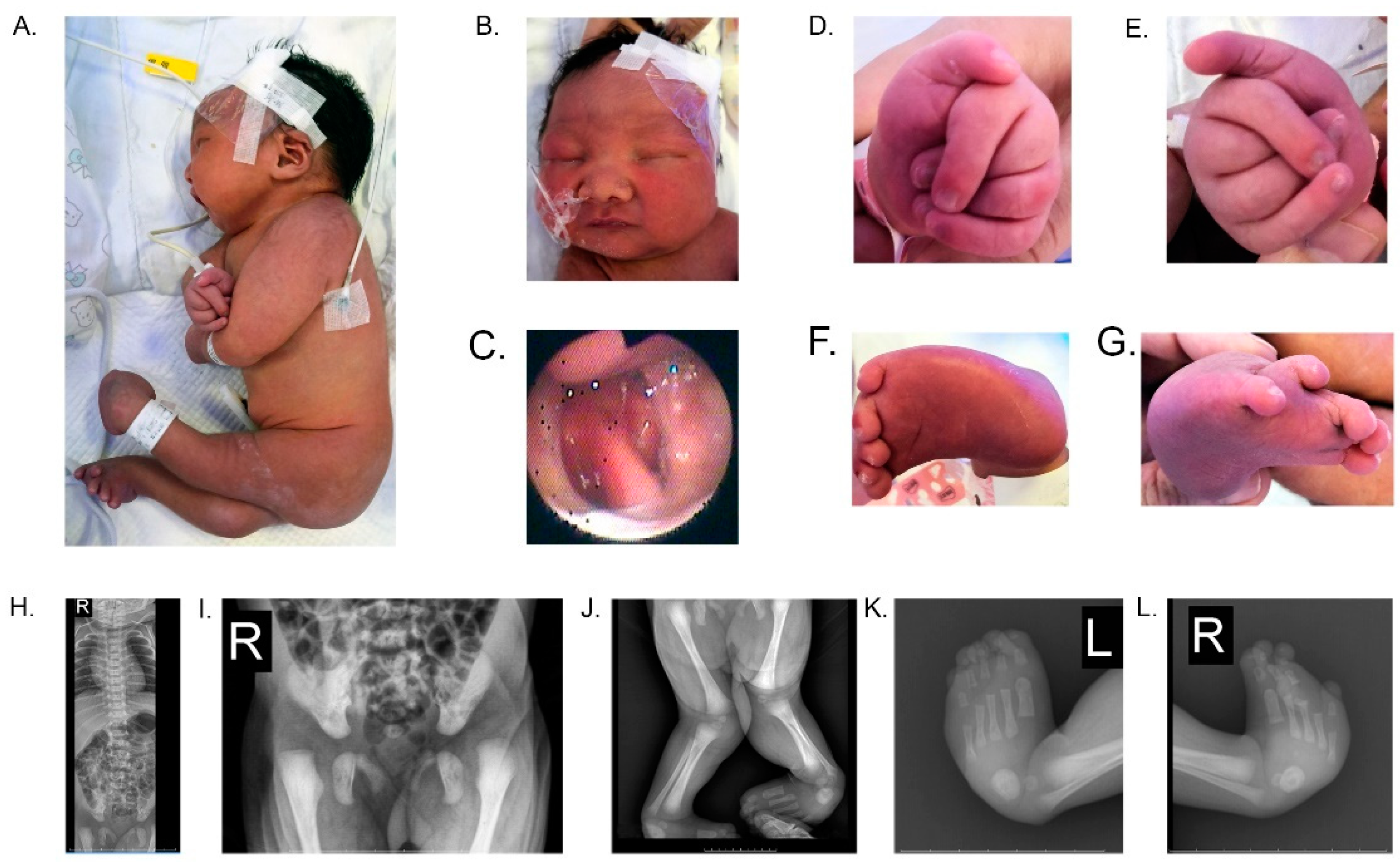
3.1. Presentation of Clinical Case
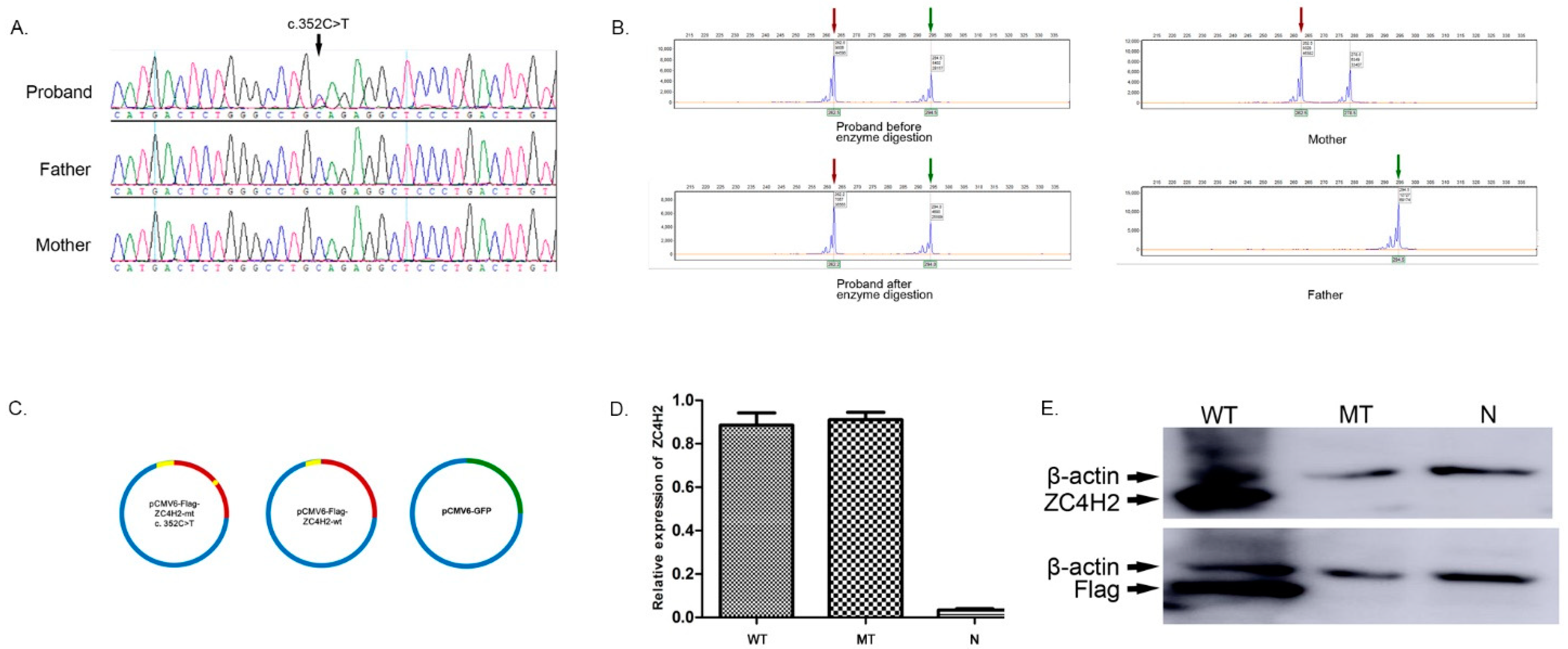
3.2. Identification of the De Novo Nonsense Mutation (c.352C>T) in the ZC4H2 Gene
3.3. In Vitro Analysis Showing the Uncoupling of Transcription and Translation of the Mutant Gene in 293T Cells
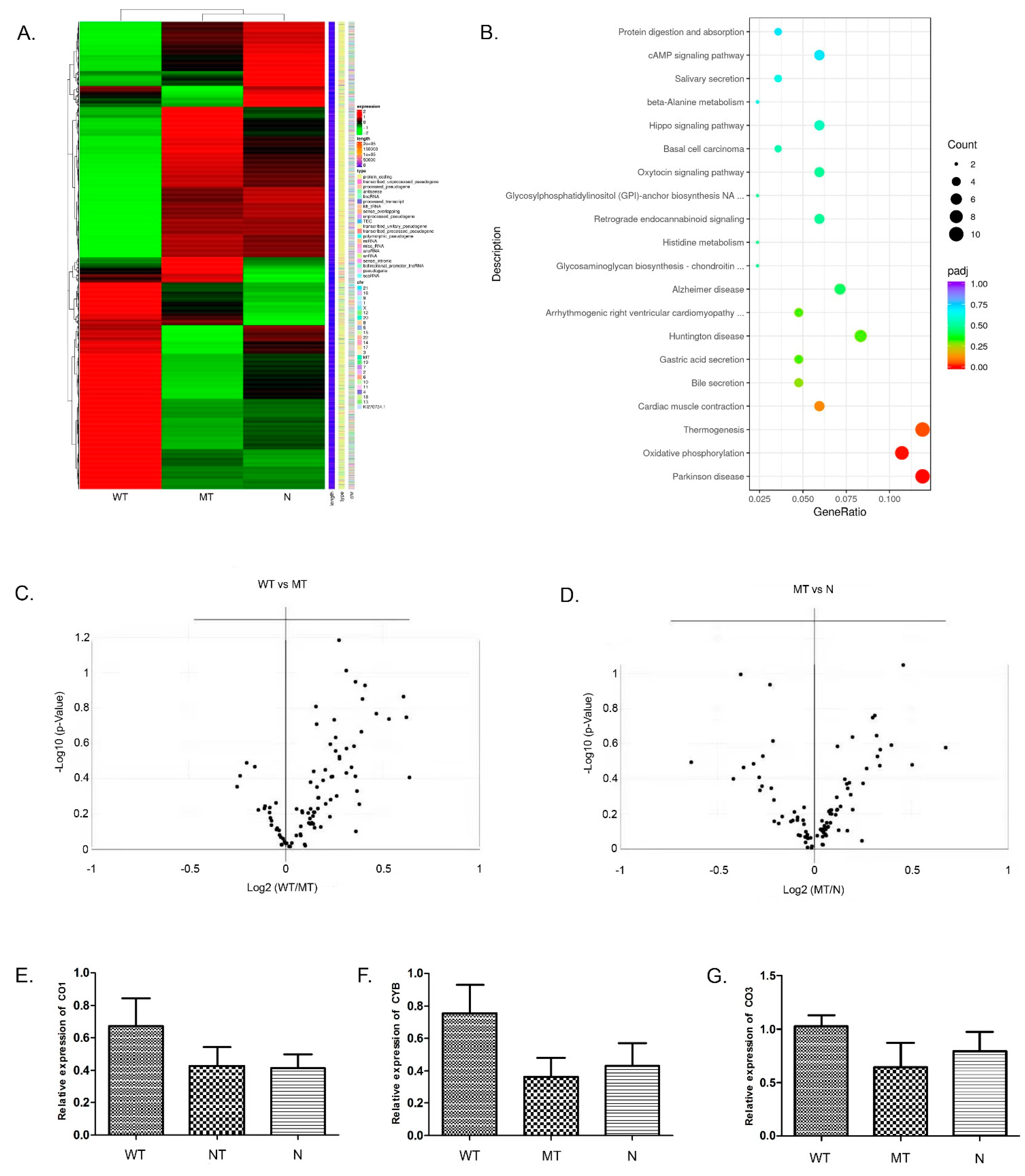
3.4. Global Transcription Profiling Using RNA-seq and Prediction of Aberrantly Regulated Pathways in the Transfected 293T Cells
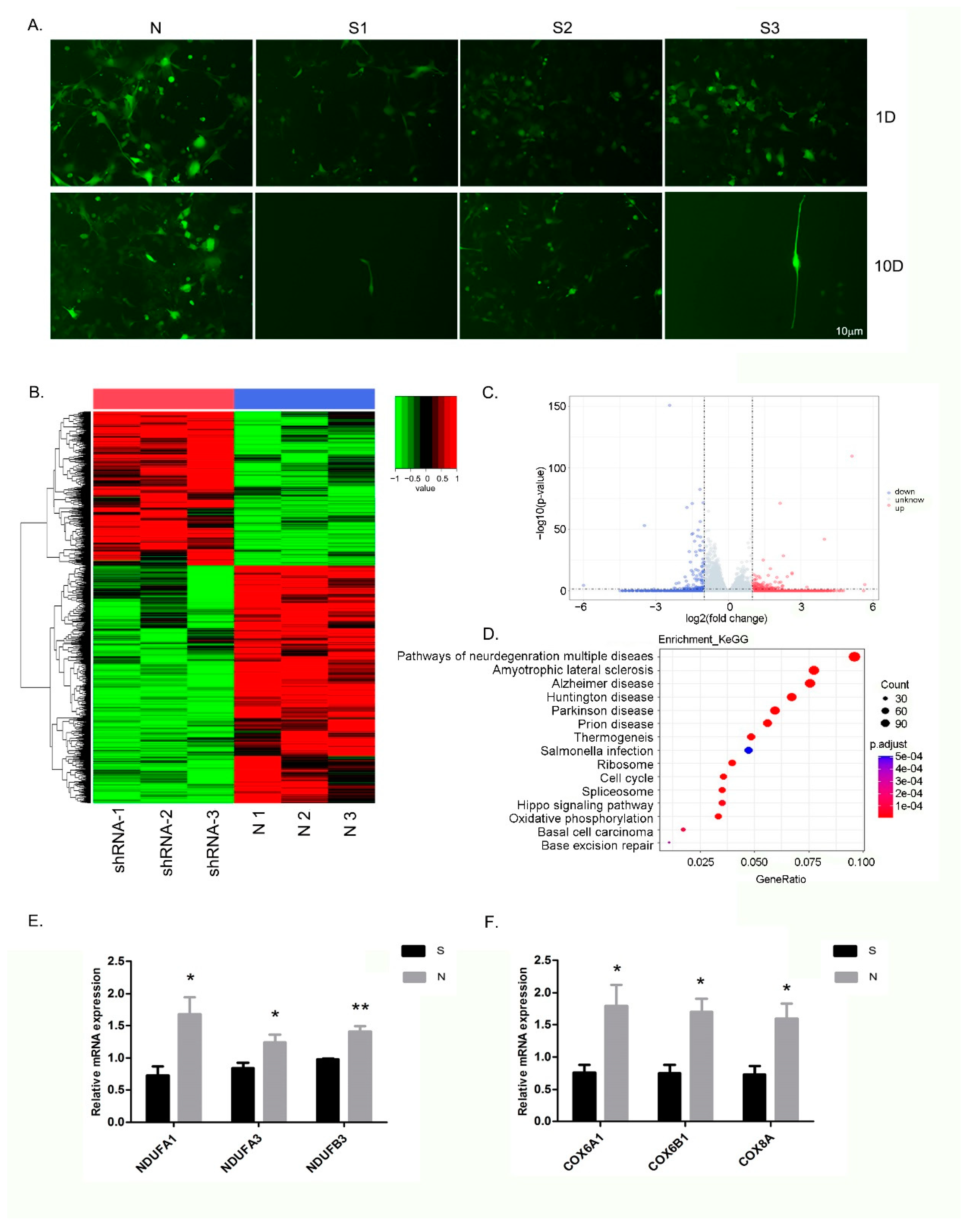
3.5. Inhibition of ZC4H2 Expression Severely Affects the Growth of NSCs
3.6. Differential Expression of Genes and Pathways in the NSCs following ZC4H2 Knockdown
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wieacker, P.; Wolff, G.; Wienker, T.F.; Sauer, M. A new X-linked syndrome with muscle atrophy, congenital contractures, and oculomotor apraxia. Am. J. Med. Genet. 1985, 20, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Nanda, I.; Van Riesen, A.; McMichael, G.; Hu, H.; Hambrock, M.; Papon, M.A.; Fischer, U.; Marouillat, S.; Ding, C. ZC4H2 mutations are associated with arthrogryposis multiplex congenita and intellectual disability through impairment of central and peripheral synaptic plasticity. Am. J. Hum. Genet. 2013, 92, 681–695. [Google Scholar] [CrossRef] [PubMed]
- May, M.; Hwang, K.S.; Miles, J.; Williams, C.; Niranjan, T.; Kahler, S.G.; Chiurazzi, P.; Steindl, K.; Van Der Spek, P.J.; Swagemakers, S. ZC4H2, an XLID gene, is required for the generation of a specific subset of CNS interneurons. Hum. Mol. Genet. 2015, 24, 4848–4861. [Google Scholar] [CrossRef] [PubMed]
- Frints, S.G.; Hennig, F.; Colombo, R.; Jacquemont, S.; Terhal, P.; Zimmerman, H.H.; Hunt, D.; Mendelsohn, B.A.; Kordaß, U.; Webster, R. Deleterious de novo variants of X-linked ZC4H2 in females cause a variable phenotype with neurogenic arthrogryposis multiplex congenita. Hum. Mutat. 2019, 40, 2270–2285. [Google Scholar] [CrossRef]
- Godfrey, N.D.; Dowlatshahi, S.; Martin, M.M.; Rothkopf, D.M. Wieacker–Wolff syndrome with associated cleft palate in a female case. Am. J. Med. Genet. Part A 2018, 176, 167–170. [Google Scholar] [CrossRef]
- Hennekam, R.C.; Barth, P.G.; Van Lookeren Campagne, W.; De Visser, M.; Dingemans, K.P. A family with severe X-linked arthrogryposis. Eur. J. Pediatrics 1991, 150, 656–660. [Google Scholar] [CrossRef]
- Zanzottera, C.; Milani, D.; Alfei, E.; Rizzo, A.; D’Arrigo, S.; Esposito, S.; Pantaleoni, C. ZC4H2 deletions can cause severe phenotype in female carriers. Am. J. Med. Genet. Part A 2017, 173, 1358–1363. [Google Scholar] [CrossRef]
- Okubo, Y.; Endo, W.; Inui, T.; Suzuki-Muromoto, S.; Miyabayashi, T.; Togashi, N.; Sato, R.; Arai-Ichinoi, N.; Kikuchi, A.; Kure, S. A severe female case of arthrogryposis multiplex congenita with brain atrophy, spastic quadriplegia and intellectual disability caused by ZC4H2 mutation. Brain Dev. 2018, 40, 334–338. [Google Scholar] [CrossRef]
- Wang, D.; Hu, D.; Guo, Z.; Hu, R.; Wang, Q.; Liu, Y.; Liu, M.; Meng, Z.; Yang, H.; Zhang, Y.; et al. A novel de novo nonsense mutation in ZC4H2 causes Wieacker-Wolff Syndrome. Mol. Genet. Genom. Med. 2020, 8, e1100. [Google Scholar] [CrossRef]
- Godfrey, D.; Torres, A.; Heidary, G.; Zahoor, H.; Lee, A.; Berry, G.; Engle, E. A 7-year old female with arthrogryposis multiplex congenita, Duane retraction syndrome, and Marcus Gunn phenomenon due to a ZC4H2 gene mutation: A clinical presentation of the Wieacker-Wolff syndrome. Ophthalmic Genet. 2021, 42, 612–614. [Google Scholar] [CrossRef]
- Deneufbourg, C.; Duquenne, A.; Biard, J.M.; Sznajer, Y. Wieacker-Wolff syndrome, a distinctive phenotype of arthrogryposis multiplex congenita caused by a “de novo” ZC4H2 gene partial deletion. Clin. Case Rep. 2021, 9, e04718. [Google Scholar] [CrossRef] [PubMed]
- Vangeel, L.; Janssens, A.; Lemmens, I.; Lievens, S.; Tavernier, J.; Voets, T. The Zinc-Finger Domain Containing Protein ZC4H2 Interacts with TRPV4, Enhancing Channel Activity and Turnover at the Plasma Membrane. Int. J. Mol. Sci. 2020, 21, 3556. [Google Scholar] [CrossRef]
- Kim, J.; Choi, T.I.; Park, S.; Kim, M.H.; Kim, C.H.; Lee, S. Rnf220 cooperates with Zc4h2 to specify spinal progenitor domains. Development 2018, 145, dev165340. [Google Scholar] [CrossRef] [PubMed]
- Song, N.N.; Ma, P.; Zhang, Q.; Zhang, L.; Wang, H.; Zhang, L.; Zhu, L.; He, C.H.; Mao, B.; Ding, Y.Q. Rnf220/Zc4h2-mediated monoubiquitylation of Phox2 is required for noradrenergic neuron development. Development 2020, 147, dev185199. [Google Scholar] [CrossRef] [PubMed]
- Skierka, A.S.; Michels, K.B. Ethical principles and placebo-controlled trials—Interpretation and implementation of the Declaration of Helsinki’s placebo paragraph in medical research. BMC Med. Ethics 2018, 19, 24. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.B.; Li, Z.; Liu, Z.; Jiang, Y.; Cai, X.B.; Wu, J. Identification of de novo germline mutations and causal genes for sporadic diseases using trio-based whole-exome/genome sequencing. Biol. Rev. Camb. Philos. Soc. 2018, 93, 1014–1031. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kubota, T.; Nonoyama, S.; Tonoki, H.; Masuno, M.; Imaizumi, K.; Kojima, M.; Wakui, K.; Shimadzu, M.; Fukushima, Y. A new assay for the analysis of X-chromosome inactivation based on methylation-specific PCR. Hum. Genet. 1999, 104, 49–55. [Google Scholar] [CrossRef]
- Hunter, S.; Jones, P.; Mitchell, A.; Apweiler, R.; Attwood, T.K.; Bateman, A.; Bernard, T.; Binns, D.; Bork, P.; Burge, S.; et al. InterPro in 2011: New developments in the family and domain prediction database. Nucleic Acids Res. 2012, 40, D306–D312. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Bernhofer, M.; Dallago, C.; Karl, T.; Satagopam, V.; Heinzinger, M.; Littmann, M.; Olenyi, T.; Qiu, J.; Schütze, K.; Yachdav, G.; et al. PredictProtein—Predicting Protein Structure and Function for 29 Years. Nucleic Acids Res. 2021, 49, W535–W540. [Google Scholar] [CrossRef]
- Qiu, J.; Bernhofer, M.; Heinzinger, M.; Kemper, S.; Norambuena, T.; Melo, F.; Rost, B. ProNA2020 predicts protein-DNA, protein-RNA, and protein-protein binding proteins and residues from sequence. J. Mol. Biol. 2020, 432, 2428–2443. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.H.; Carpenter, N.J. Unique X-linked mental retardation syndrome with fingertip arches and contractures linked to Xq21.31. Am. J. Med. Genet. 1991, 38, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Kondo, D.; Noguchi, A.; Takahashi, I.; Kubota, H.; Yano, T.; Sato, Y.; Toyono, M.; Sawaishi, Y.; Takahashi, T. A novel ZC4H2 gene mutation, K209N, in Japanese siblings with arthrogryposis multiplex congenita and intellectual disability: Characterization of the K209N mutation and clinical findings. Brain Dev. 2018, 40, 760–767. [Google Scholar] [CrossRef]
- Nagara, S.; Fukaya, S.; Muramatsu, Y.; Kaname, T.; Tanaka, T. A case report of rare ZC4H2-associated disorders associated with three large hernias. Pediatr. Int. 2020, 62, 985–986. [Google Scholar] [CrossRef]
- Piccolo, G.; d’Annunzio, G.; Amadori, E.; Riva, A.; Borgia, P.; Tortora, D.; Maghnie, M.; Minetti, C.; Gitto, E.; Iacomino, M.; et al. Neuromuscular and Neuroendocrinological Features Associated with ZC4H2-Related Arthrogryposis Multiplex Congenita in a Sicilian Family: A Case Report. Front. Neurol. 2021, 12, 704747. [Google Scholar] [CrossRef]
- Vacca, M.; Della Ragione, F.; Scalabrì, F.; D’Esposito, M. X inactivation and reactivation in X-linked diseases. Semin. Cell Dev. Biol. 2016, 56, 78–87. [Google Scholar] [CrossRef]
- Arrázola, M.S.; Andraini, T.; Szelechowski, M.; Mouledous, L.; Arnauné-Pelloquin, L.; Davezac, N.; Belenguer, P.; Rampon, C.; Miquel, M.C. Mitochondria in Developmental and Adult Neurogenesis. Neurotox. Res. 2019, 36, 257–267. [Google Scholar] [CrossRef]
- Nicaise, A.M.; Willis, C.M.; Crocker, S.J.; Pluchino, S. Stem Cells of the Aging Brain. Front. Aging Neurosci. 2020, 12, 247. [Google Scholar] [CrossRef]
- Pitceathly, R.D.; Rahman, S.; Wedatilake, Y.; Polke, J.M.; Cirak, S.; Foley, A.R.; Sailer, A.; Hurles, M.E.; Stalker, J.; Hargreaves, I.; et al. NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 2013, 3, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Abdulhag, U.N.; Soiferman, D.; Schueler-Furman, O.; Miller, C.; Shaag, A.; Elpeleg, O.; Edvardson, S.; Saada, A. Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur. J. Hum. Genet. 2015, 23, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Massa, V.; Fernandez-Vizarra, E.; Alshahwan, S.; Bakhsh, E.; Goffrini, P.; Ferrero, I.; Mereghetti, P.; D’Adamo, P.; Gasparini, P.; Zeviani, M. Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am. J. Hum. Genet. 2008, 82, 1281–1289. [Google Scholar] [CrossRef]
- Tamiya, G.; Makino, S.; Hayashi, M.; Abe, A.; Numakura, C.; Ueki, M.; Tanaka, A.; Ito, C.; Toshimori, K.; Ogawa, N. A mutation of COX6A1 causes a recessive axonal or mixed form of Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2014, 95, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Rotko, D.; Kudin, A.P.; Zsurka, G.; Kulawiak, B.; Szewczyk, A.; Kunz, W.S. Loss of the smallest subunit of cytochrome c oxidase, COX8A, causes Leigh-like syndrome and epilepsy. Brain 2016, 139, 338–345. [Google Scholar]
- Fang, J.; Uchiumi, T.; Yagi, M.; Matsumoto, S.; Amamoto, R.; Takazaki, S.; Yamaza, H.; Nonaka, K.; Kang, D. Dihydro-orotate dehydrogenase is physically associated with the respiratory complex and its loss leads to mitochondrial dysfunction. Biosci. Rep. 2013, 33, e00021. [Google Scholar] [CrossRef] [PubMed]
- Minoux, M.; Rijli, F.M. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development 2010, 137, 2605–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, J.-J.; Cai, Q.; Xu, M.; Liu, Y.-N.; Li, W.-R.; Li, J.; Ma, L.; Cai, C.; Gong, X.-H.; Zeng, Y.-T.; et al. Loss of Protein Function Causing Severe Phenotypes of Female-Restricted Wieacker Wolff Syndrome due to a Novel Nonsense Mutation in the ZC4H2 Gene. Genes 2022, 13, 1558. https://doi.org/10.3390/genes13091558
Sun J-J, Cai Q, Xu M, Liu Y-N, Li W-R, Li J, Ma L, Cai C, Gong X-H, Zeng Y-T, et al. Loss of Protein Function Causing Severe Phenotypes of Female-Restricted Wieacker Wolff Syndrome due to a Novel Nonsense Mutation in the ZC4H2 Gene. Genes. 2022; 13(9):1558. https://doi.org/10.3390/genes13091558
Chicago/Turabian StyleSun, Jing-Jing, Qin Cai, Miao Xu, Yan-Na Liu, Wan-Rui Li, Juan Li, Li Ma, Cheng Cai, Xiao-Hui Gong, Yi-Tao Zeng, and et al. 2022. "Loss of Protein Function Causing Severe Phenotypes of Female-Restricted Wieacker Wolff Syndrome due to a Novel Nonsense Mutation in the ZC4H2 Gene" Genes 13, no. 9: 1558. https://doi.org/10.3390/genes13091558
APA StyleSun, J.-J., Cai, Q., Xu, M., Liu, Y.-N., Li, W.-R., Li, J., Ma, L., Cai, C., Gong, X.-H., Zeng, Y.-T., Ren, Z.-R., & Zeng, F. (2022). Loss of Protein Function Causing Severe Phenotypes of Female-Restricted Wieacker Wolff Syndrome due to a Novel Nonsense Mutation in the ZC4H2 Gene. Genes, 13(9), 1558. https://doi.org/10.3390/genes13091558