
Application of Feature Selection and Deep Learning for Cancer Prediction Using DNA Methylation Markers



Abstract
:1. Introduction
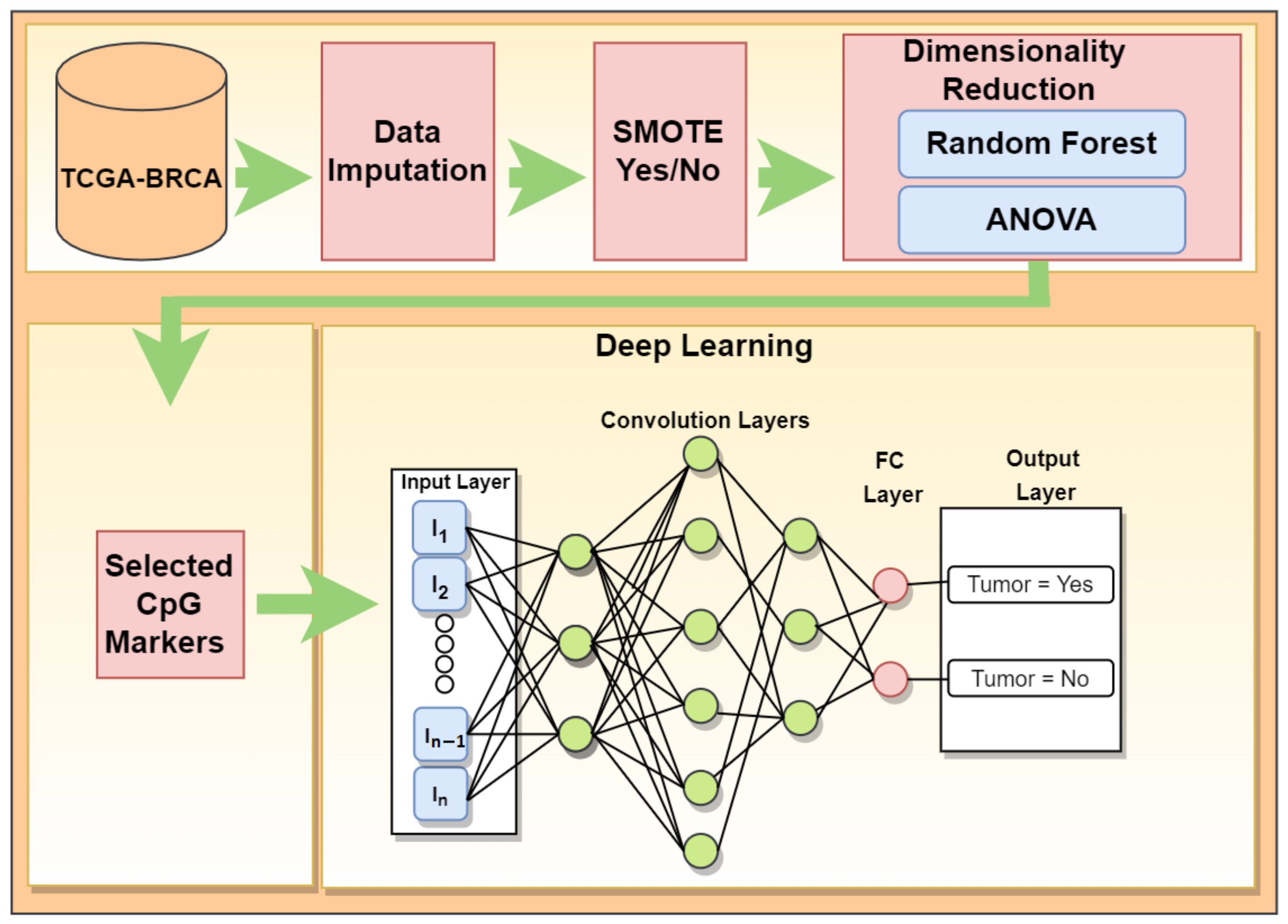
2. Materials and Methods
2.1. Dataset
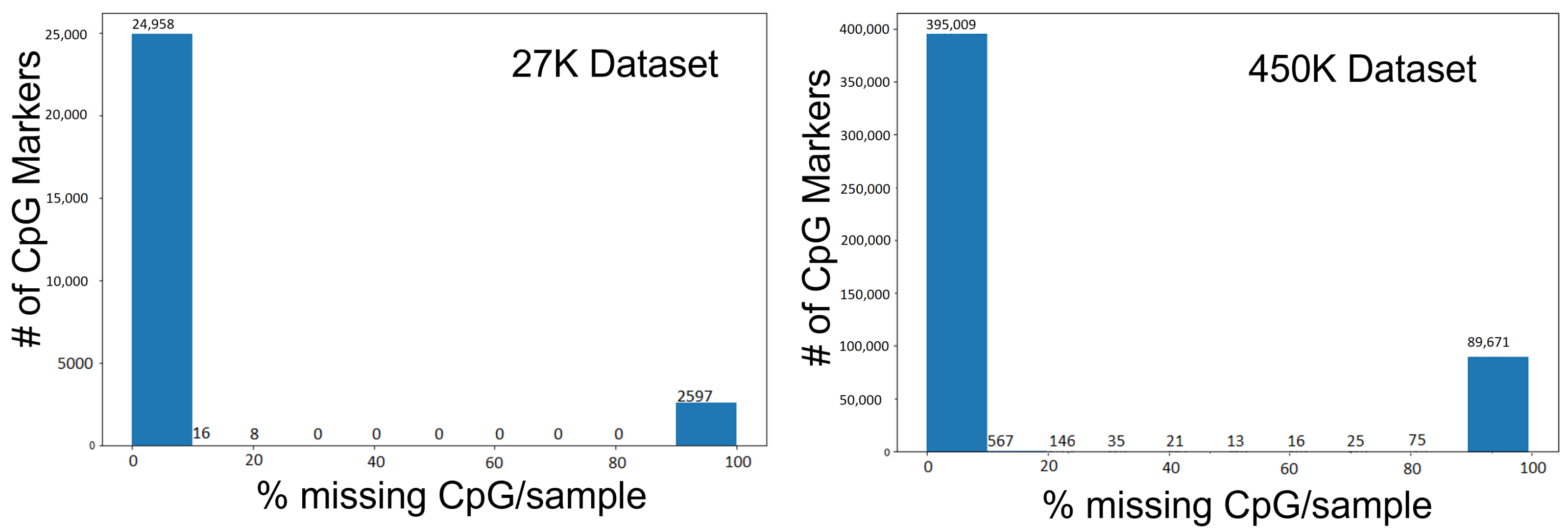
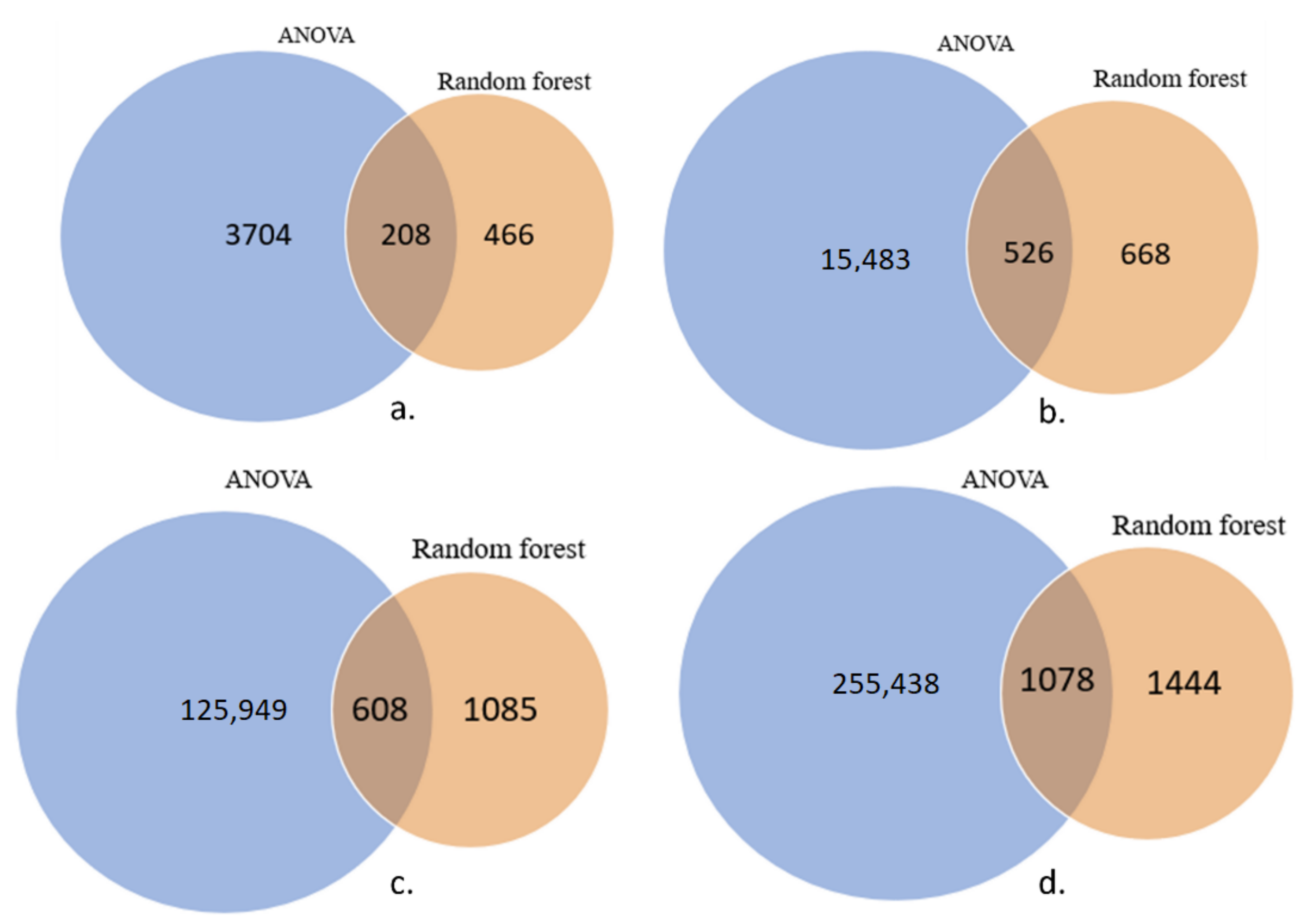
2.2. Feature Selection to Reduce Dimensionality
2.3. Handling Data Imbalance
2.4. Deep Learning Application for Cancer Prediction
- Dataset is separated into positive and negative tumor outcomes.
- The limiting outcome is randomly separated into two sets containing 70% (for training) and 30% (for testing) of the data.
- A subset of the non-limiting outcome, equal to 70% of the limiting outcome, is randomly chosen.
- The two subsets of the two outcomes, equivalent in number, is combined to form the training data.
- All remaining samples are combined to form the testing data set.
- Both data sets are randomly shuffled internally.
2.5. Gene Set Enrichment Analysis (GSEA)
2.6. Survival Analysis
3. Results
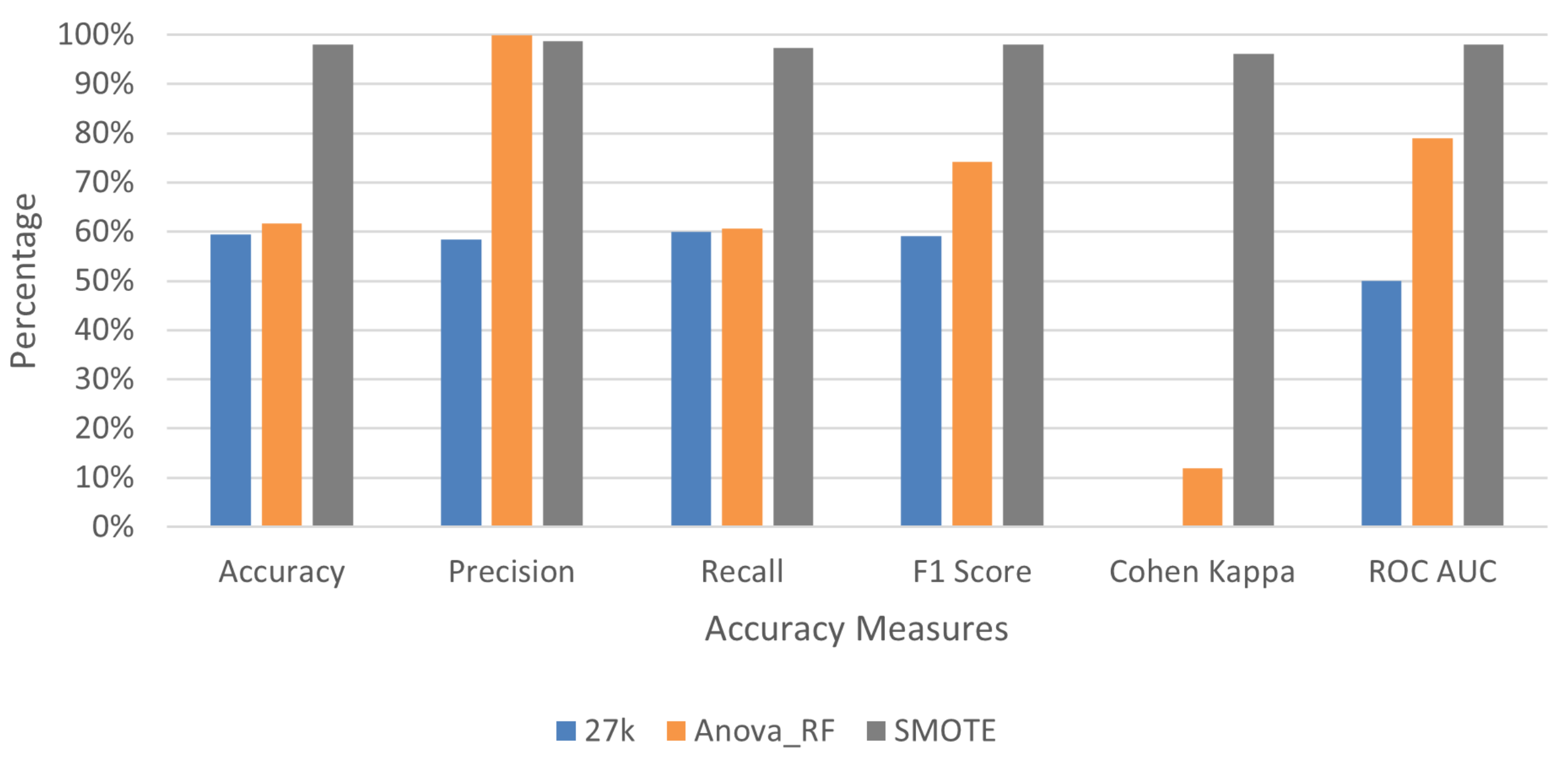
3.1. Feature Selection to Reduce Dimensionality
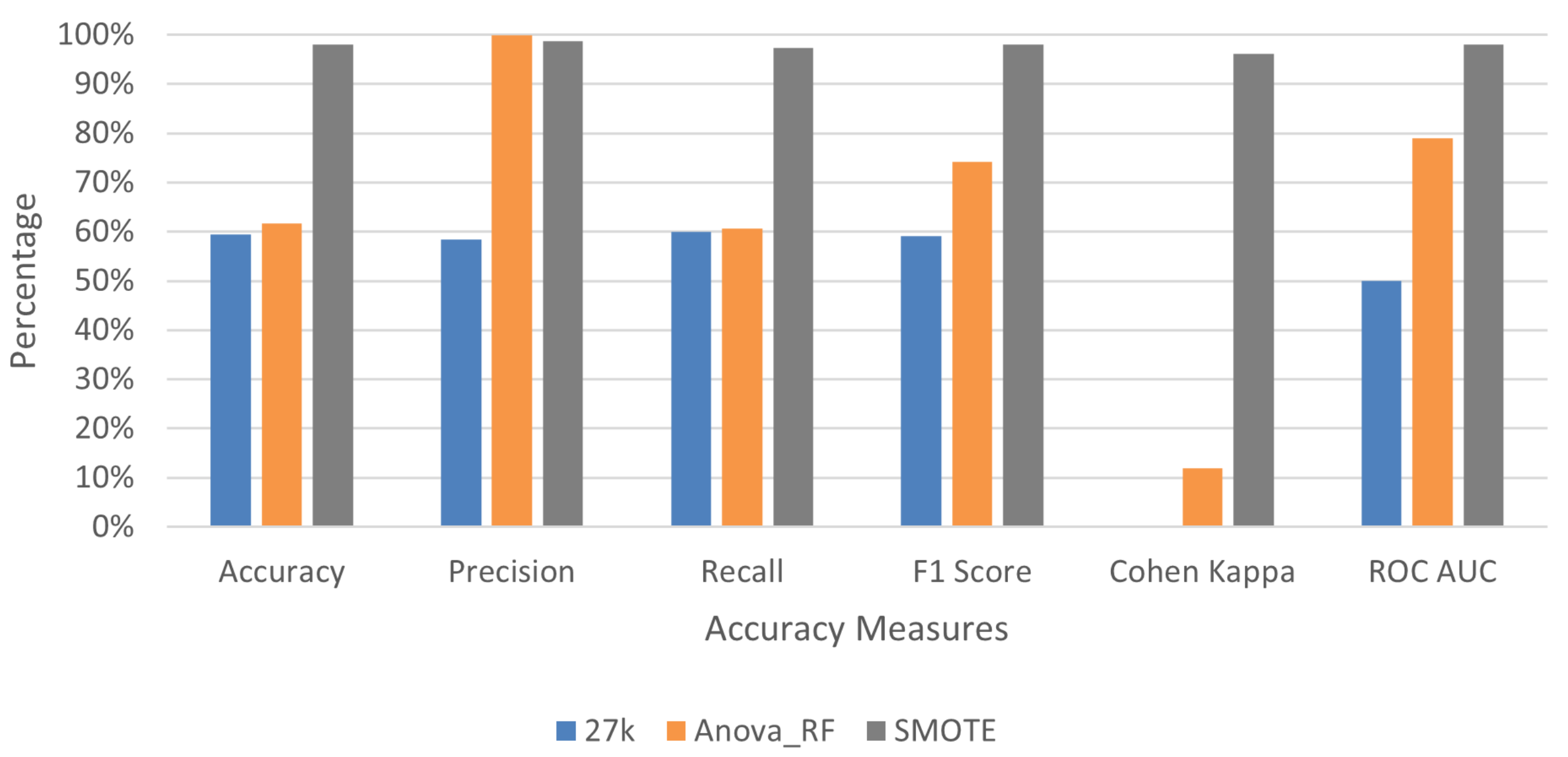
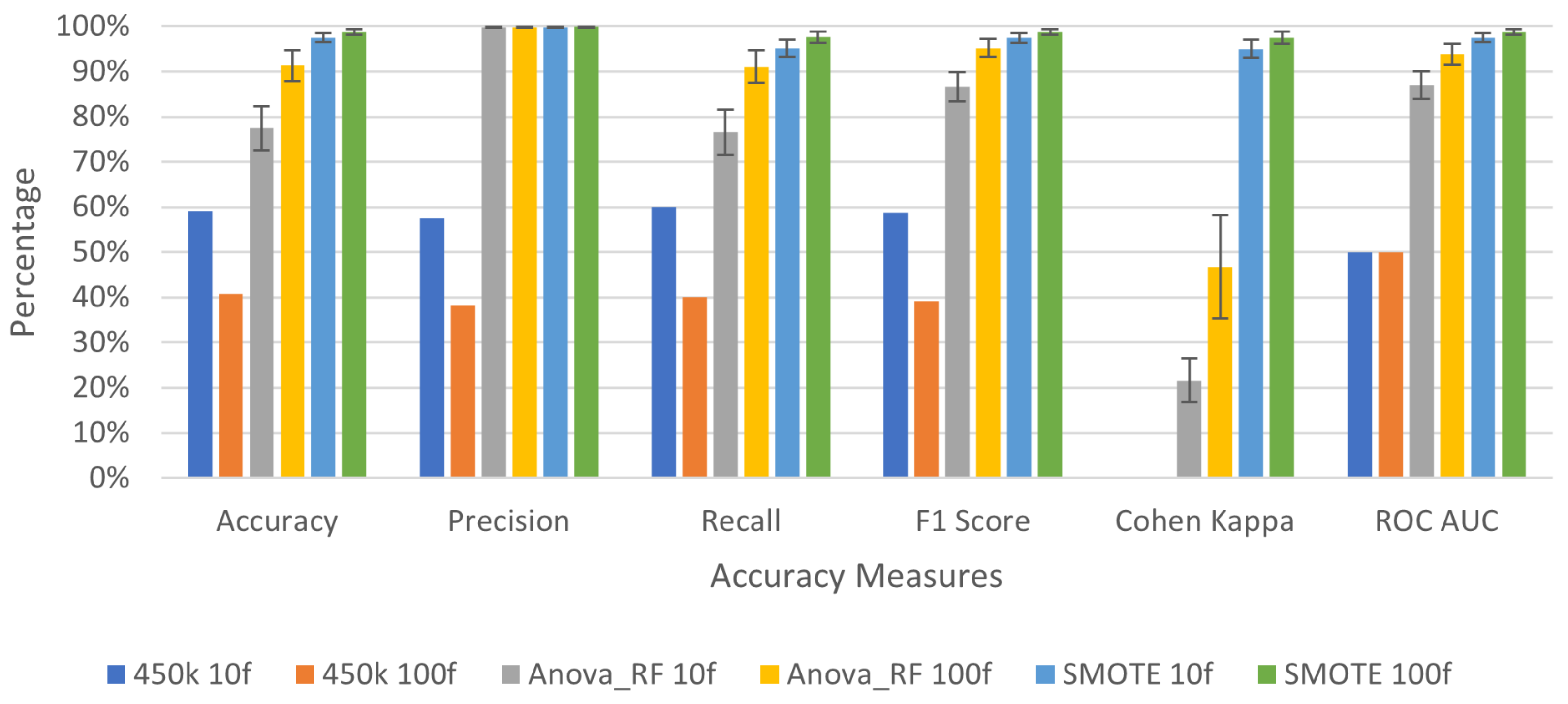
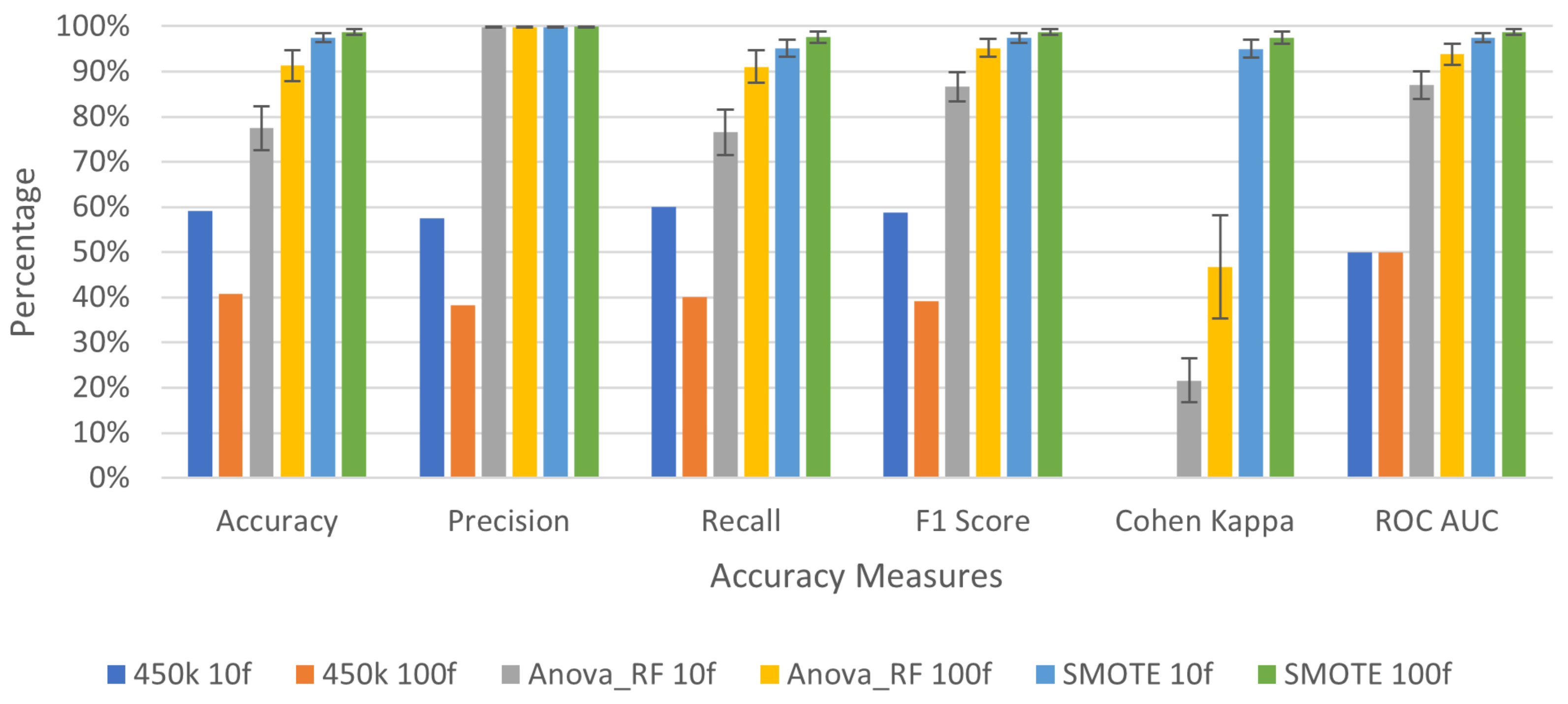
3.2. Deep Learning Application for Cancer Prediction
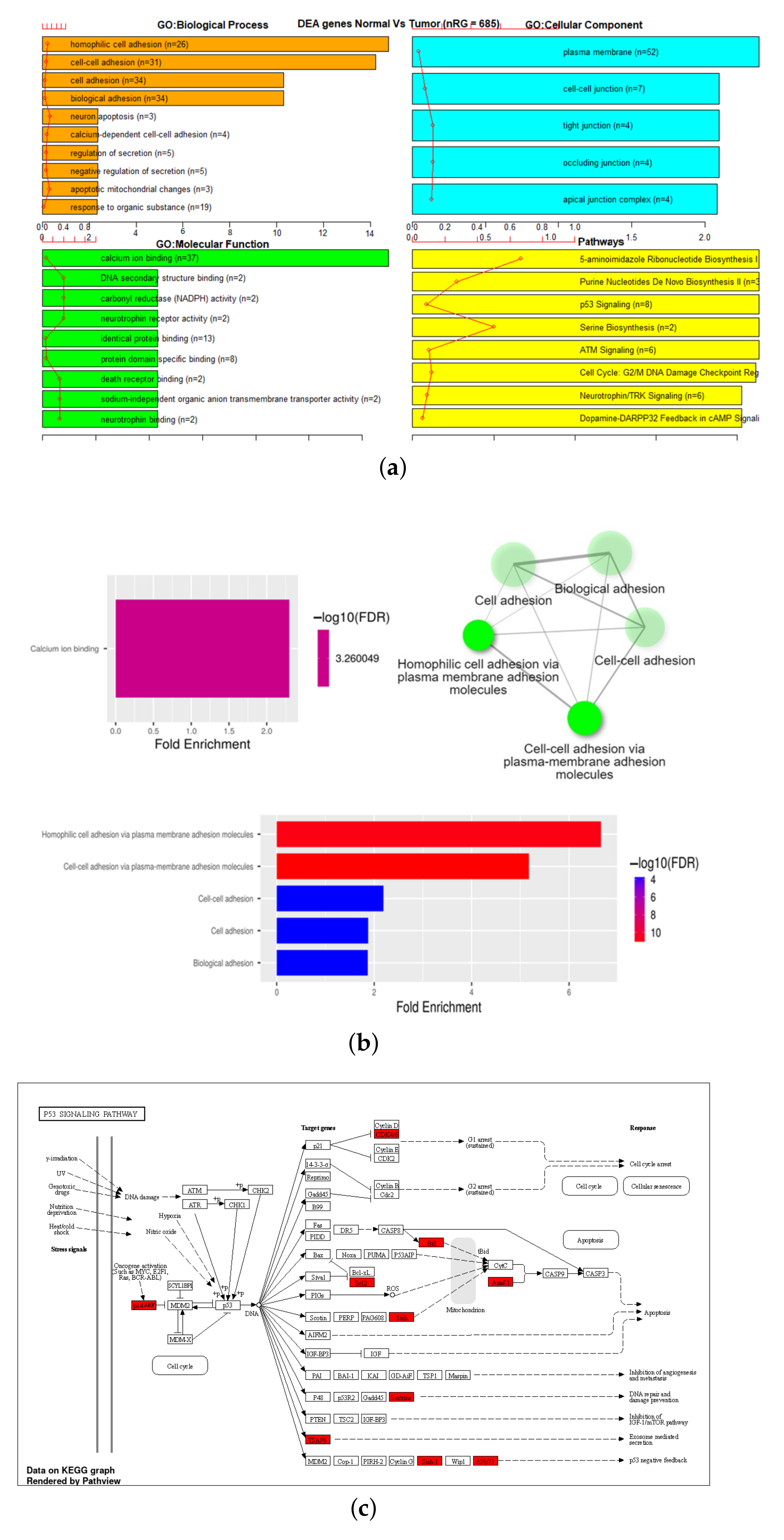
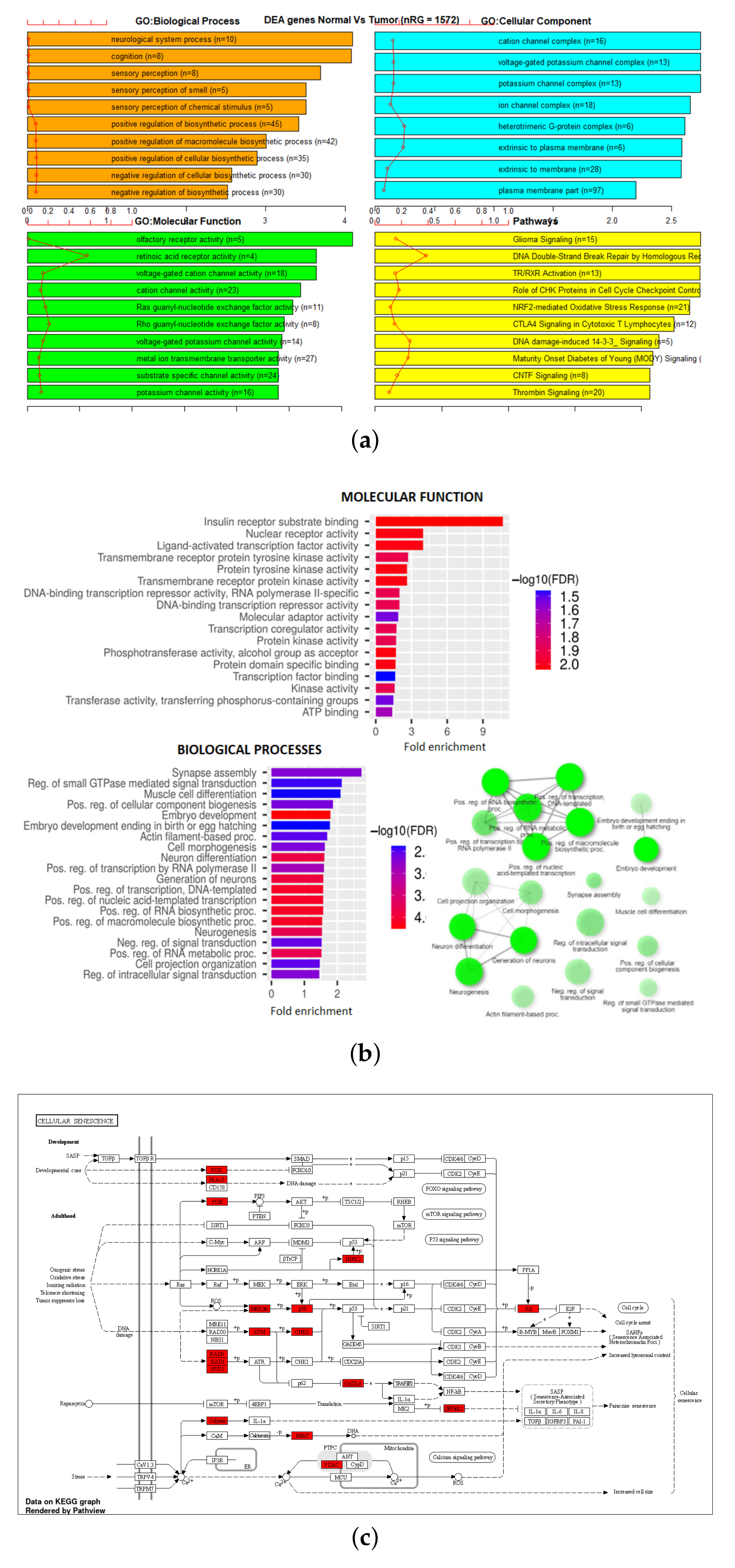
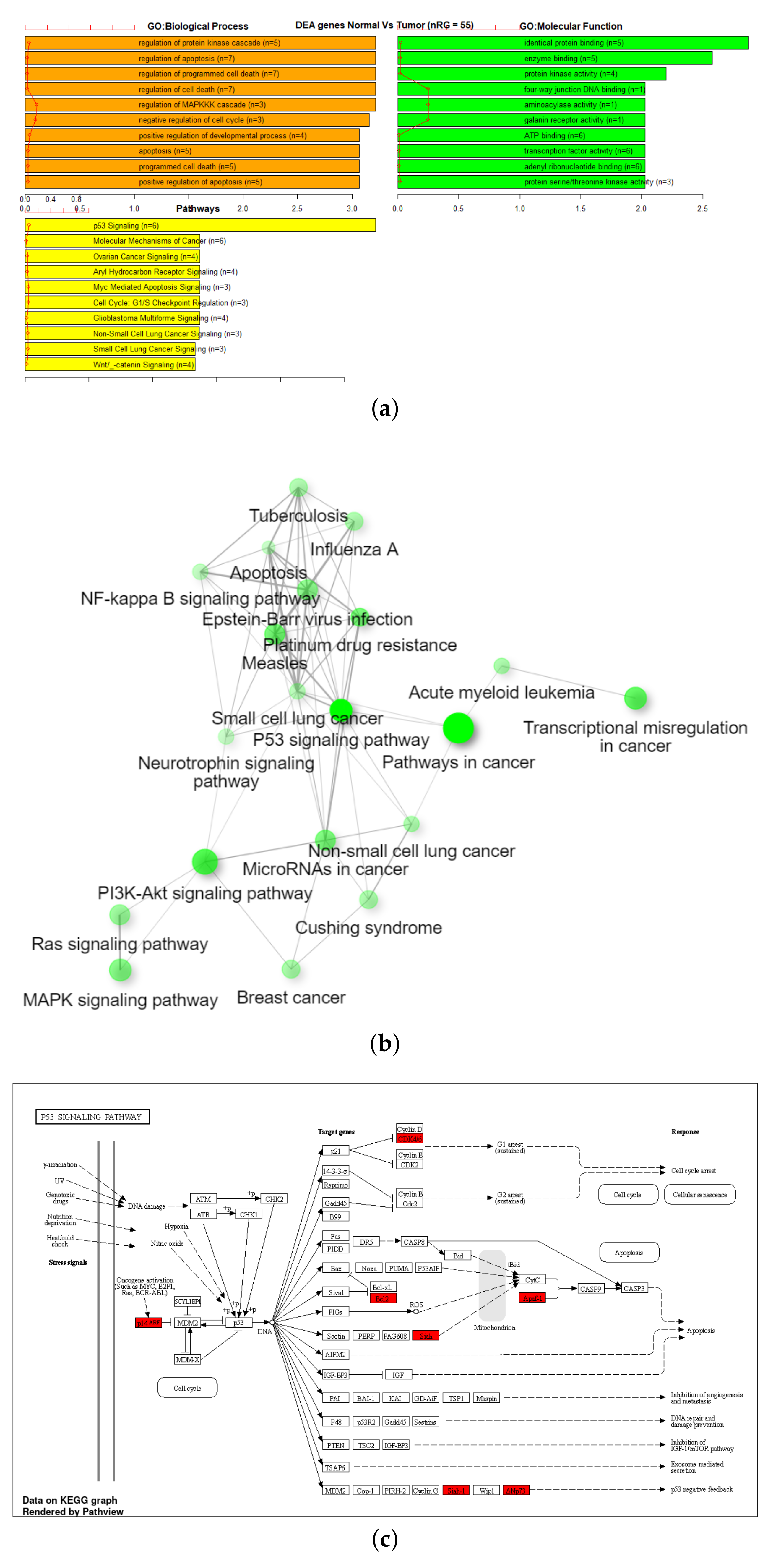
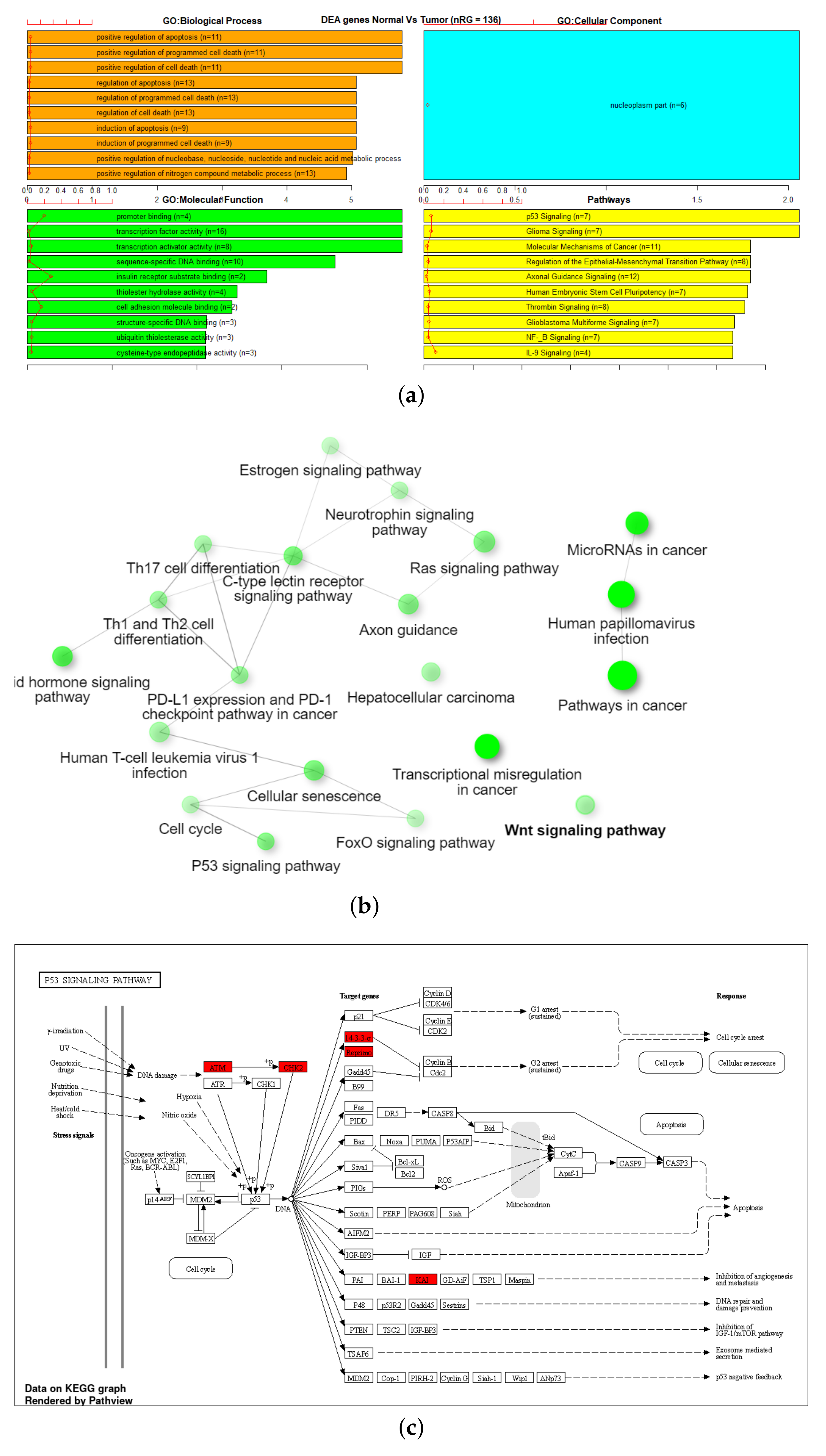
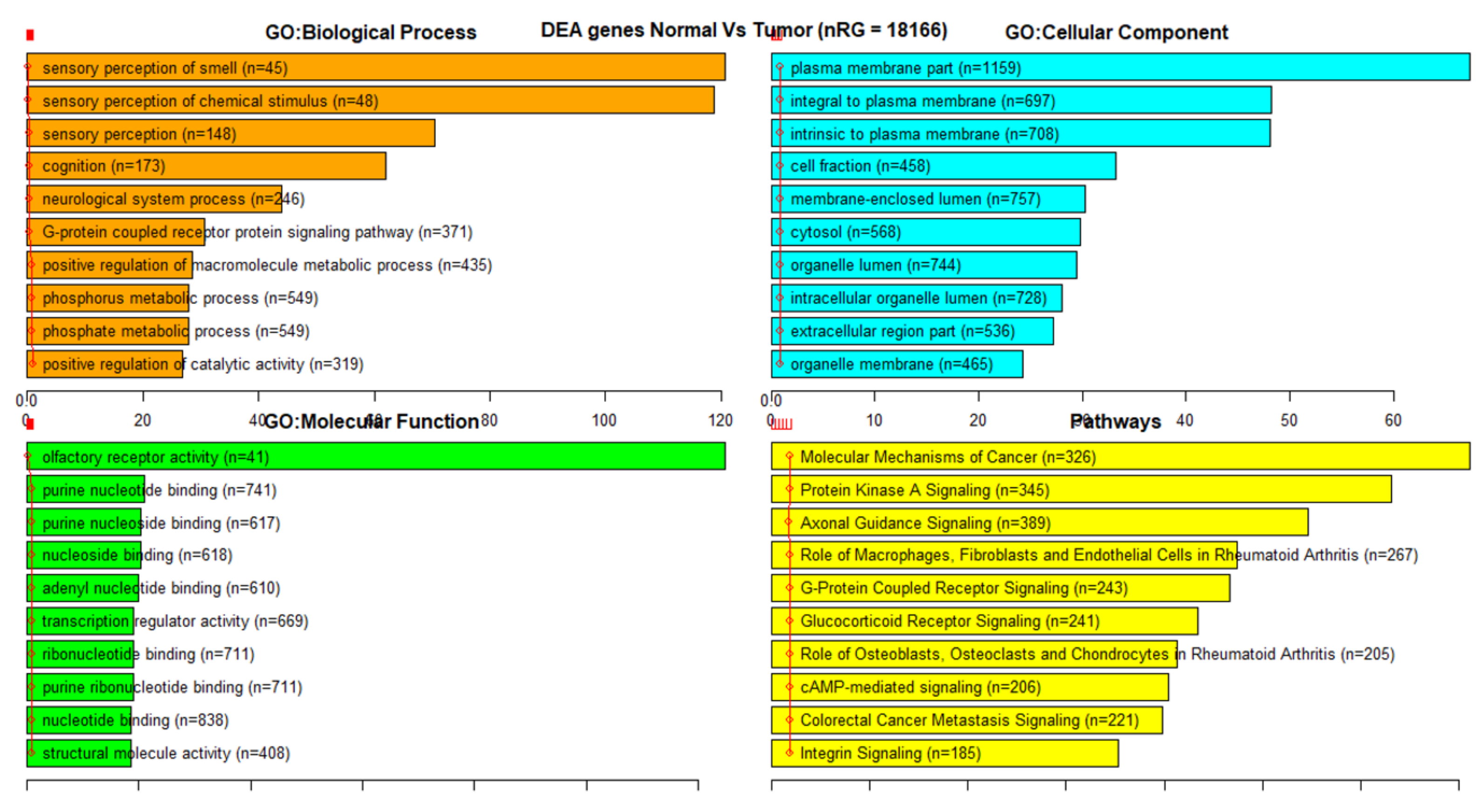
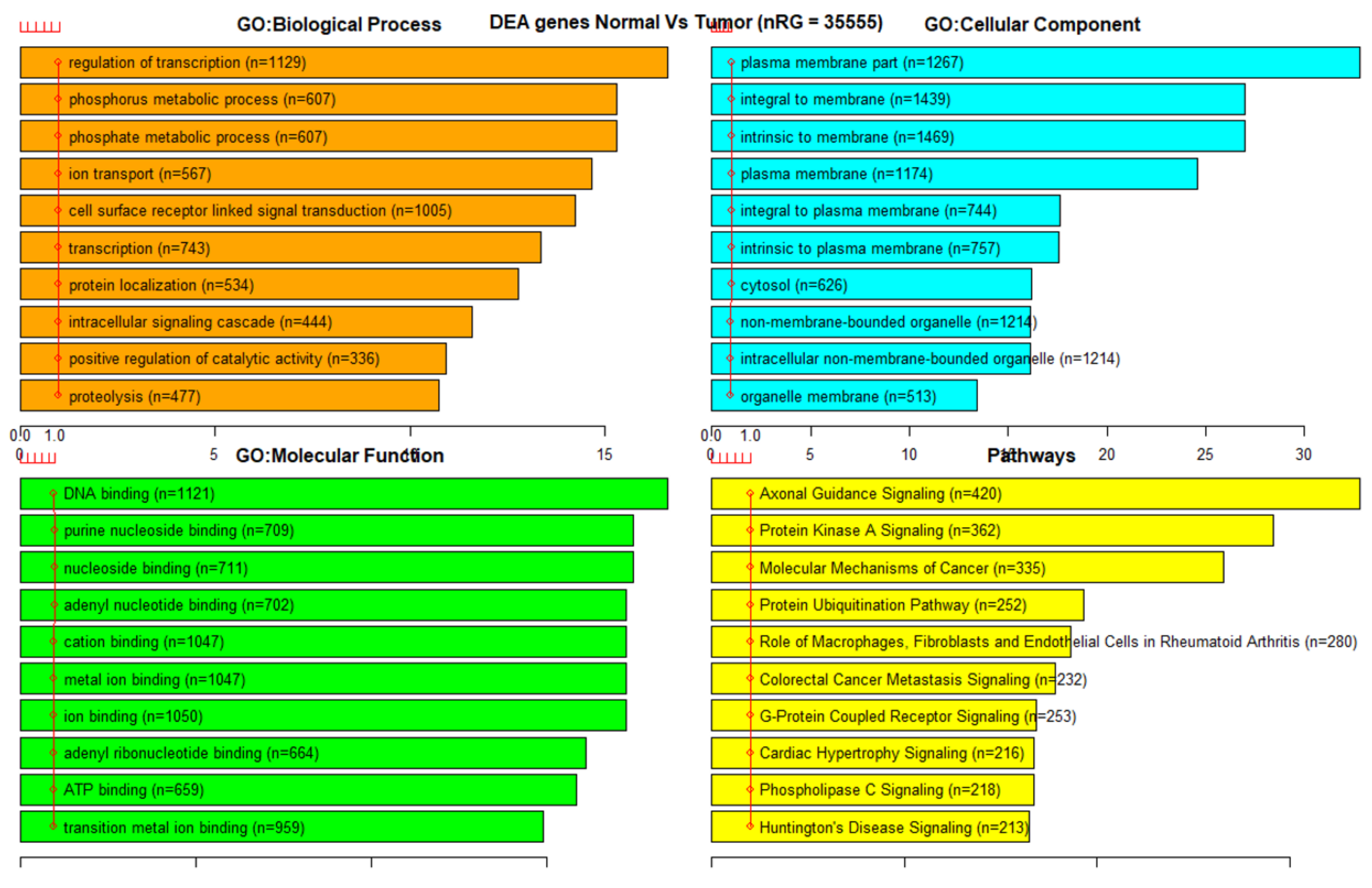
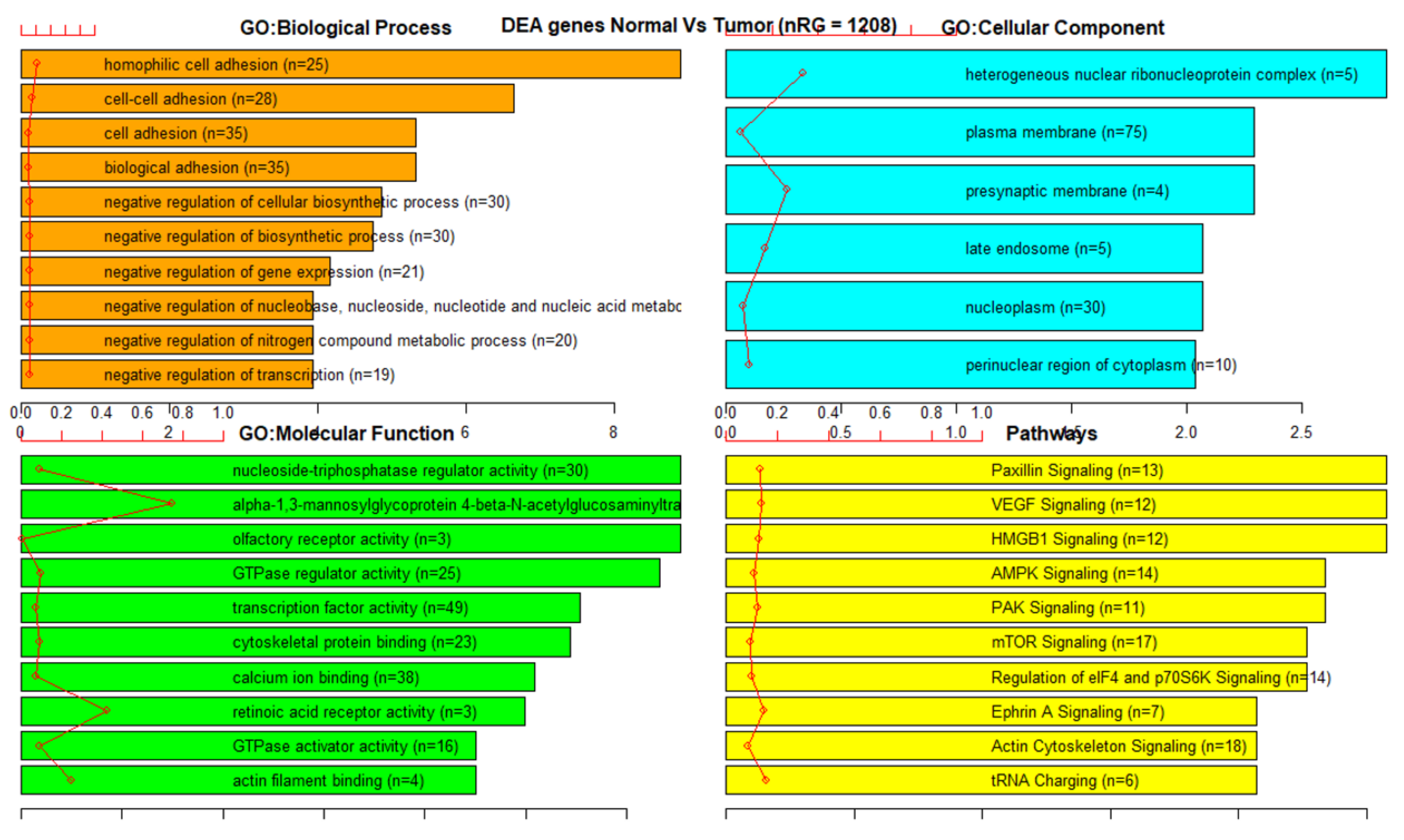
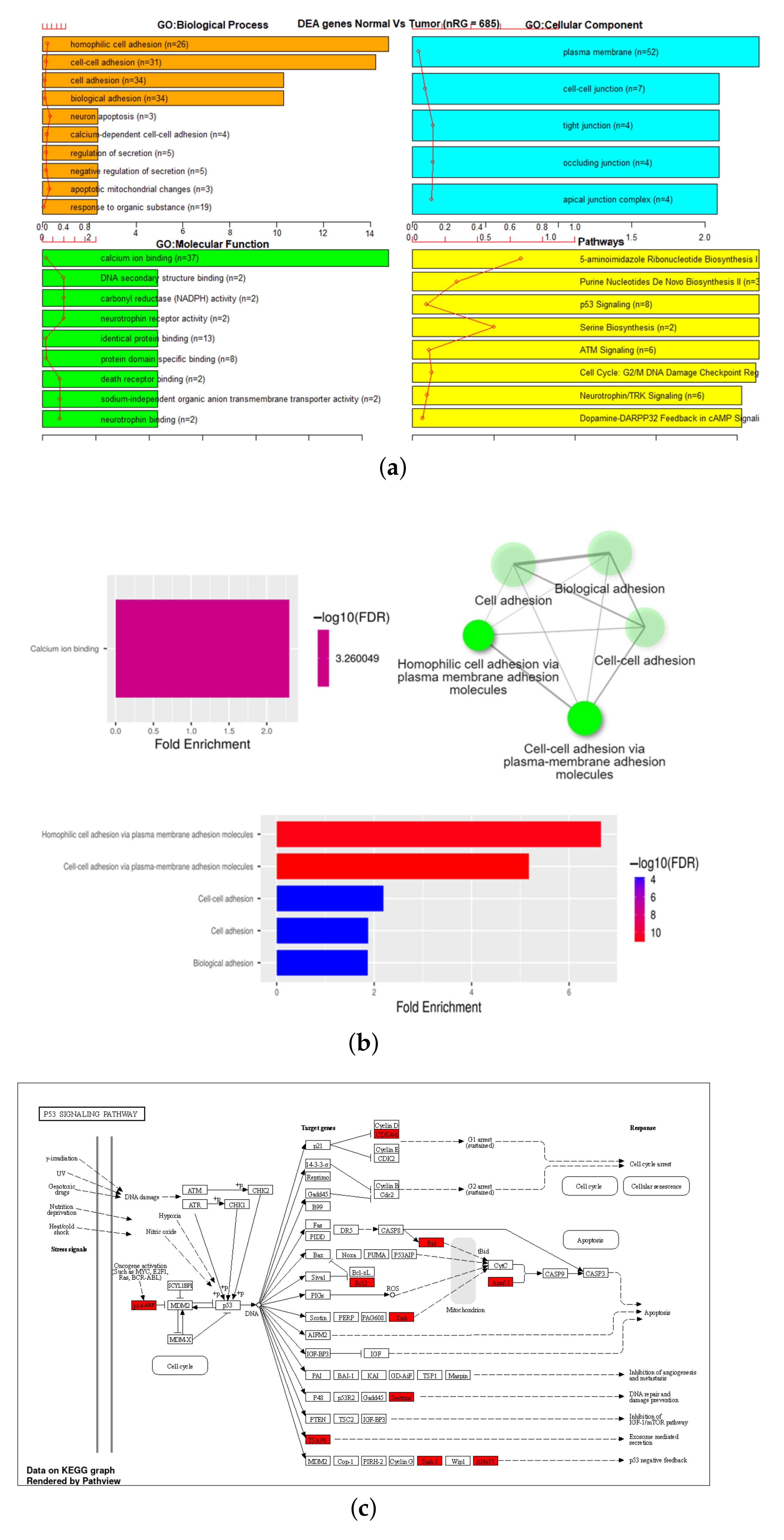
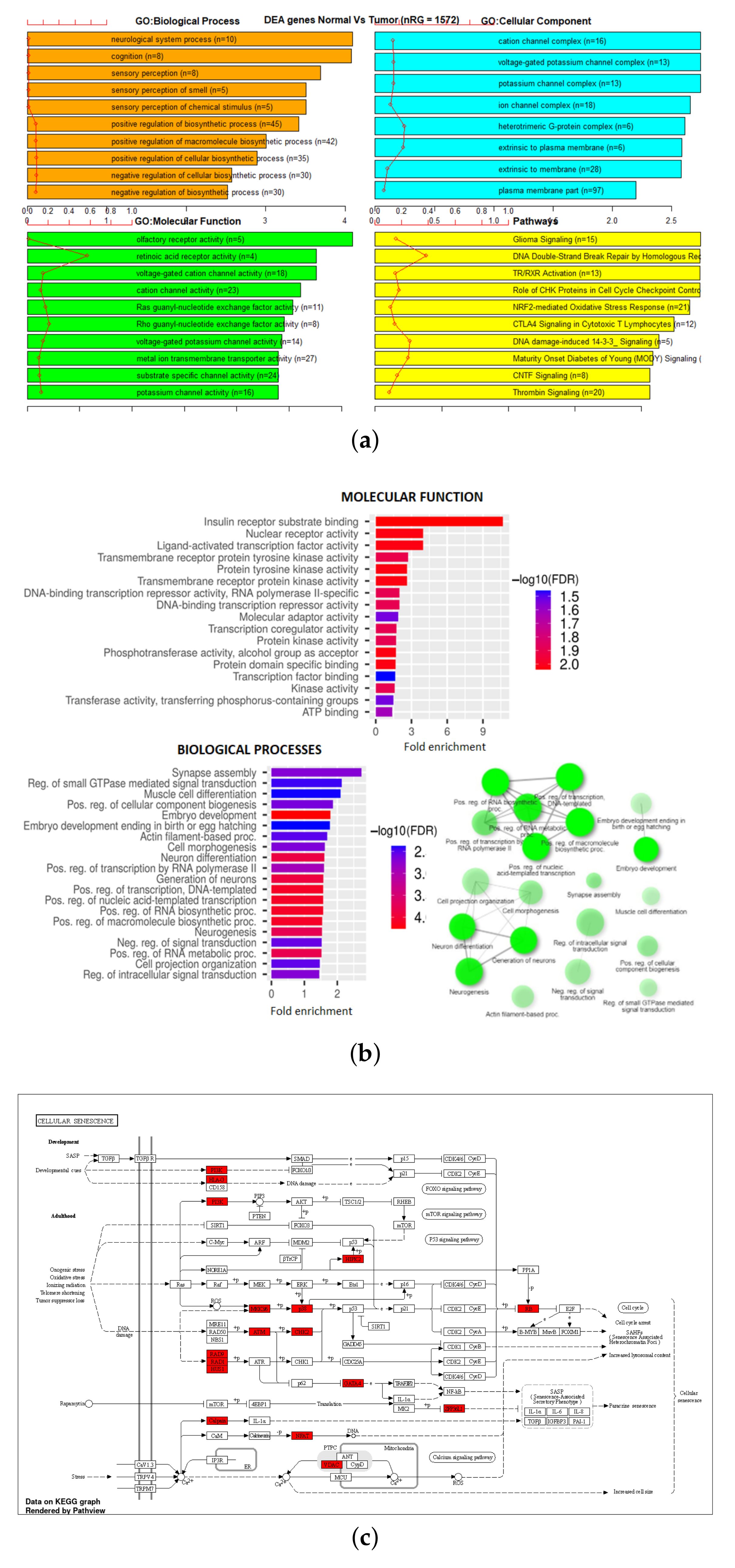
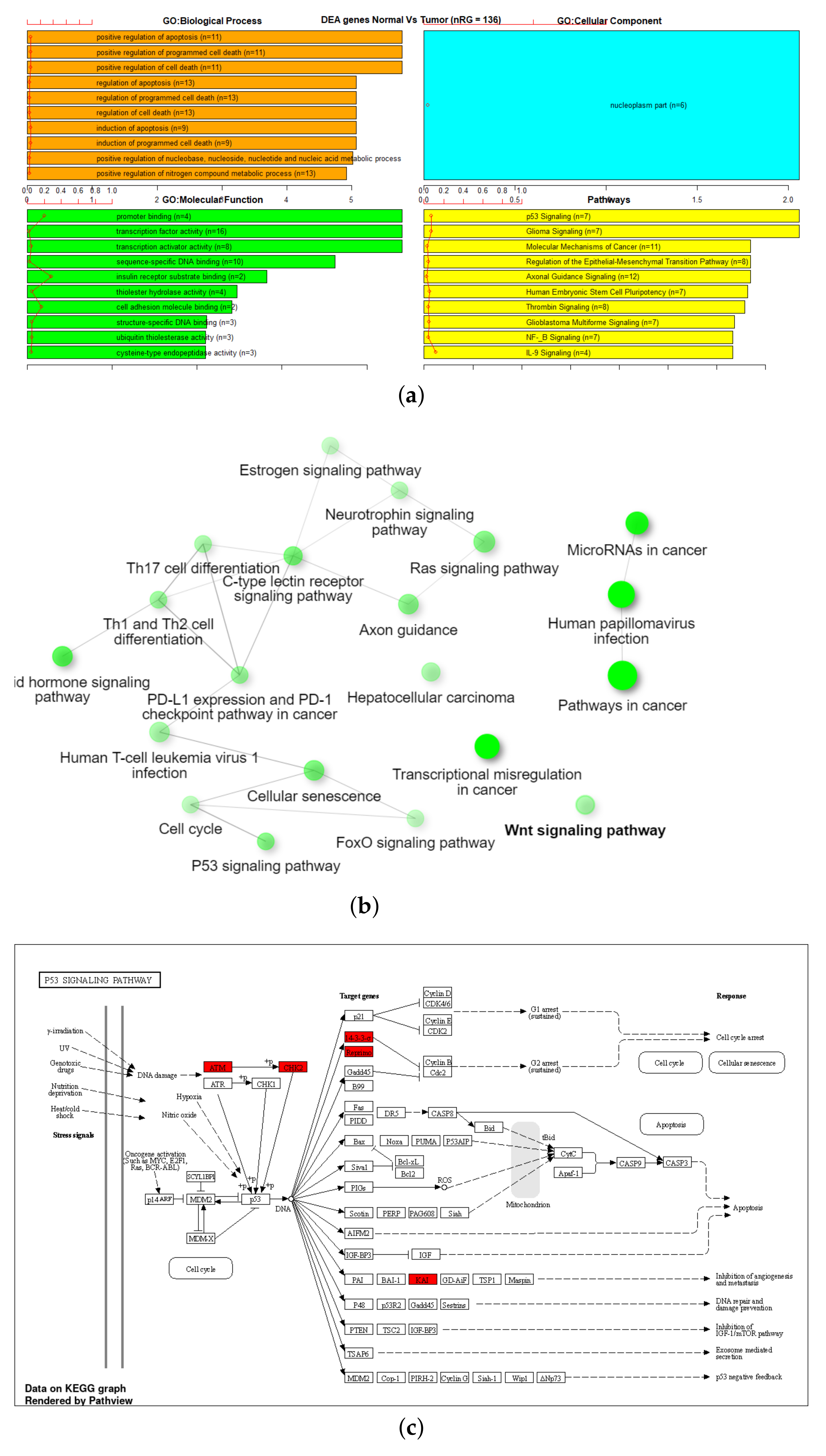
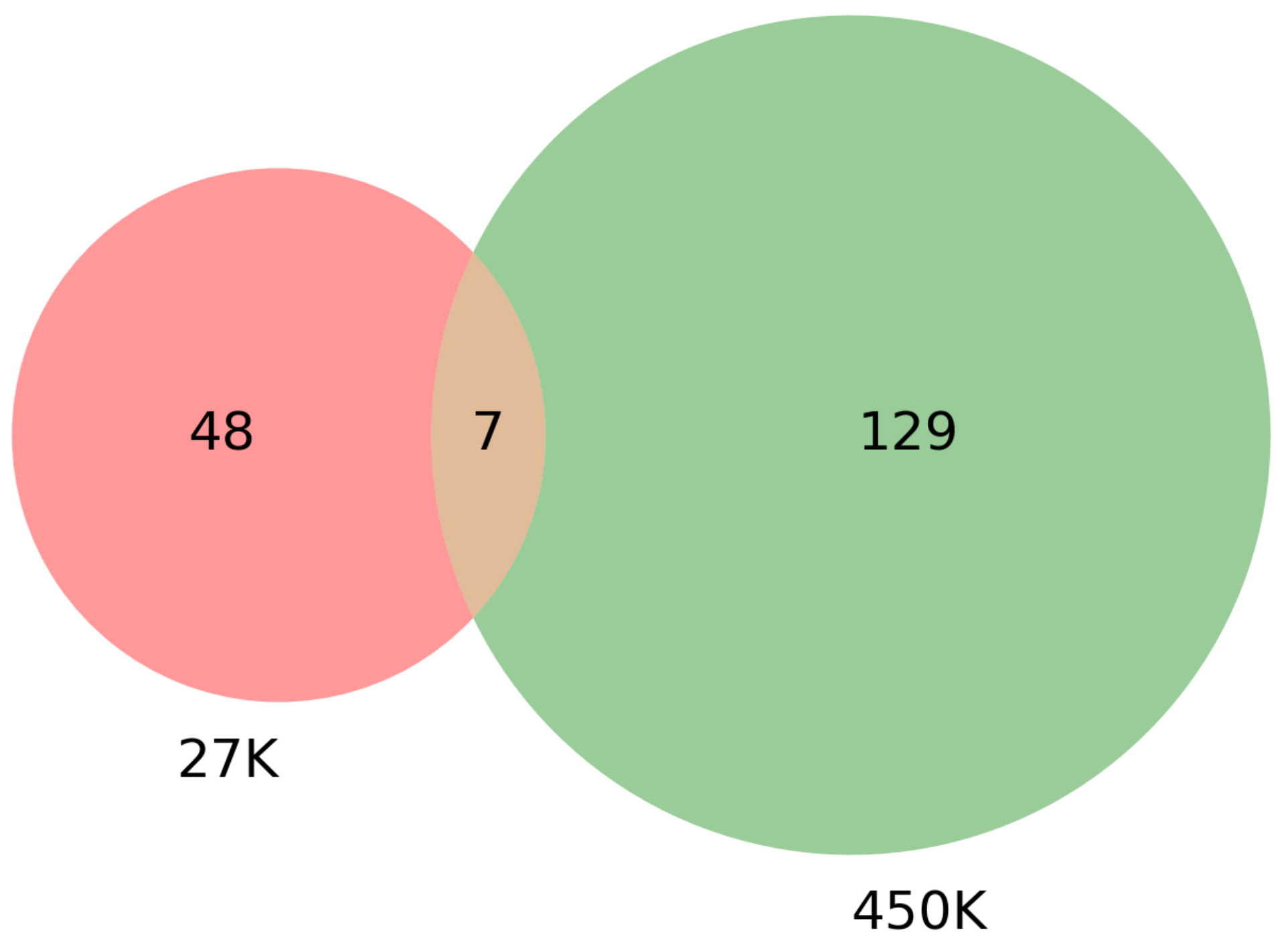
3.3. Gene Set Enrichment Analysis (GSEA)
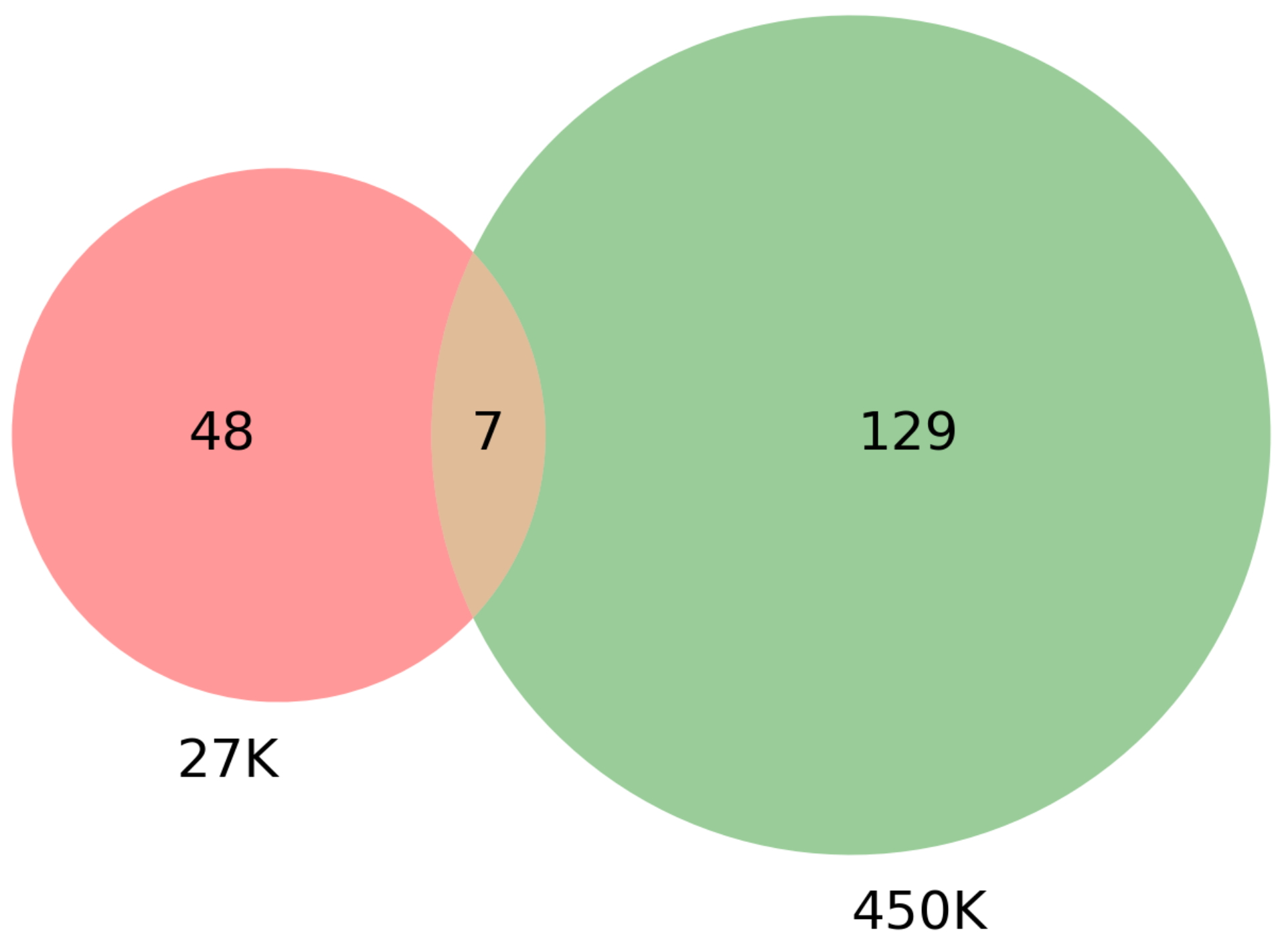
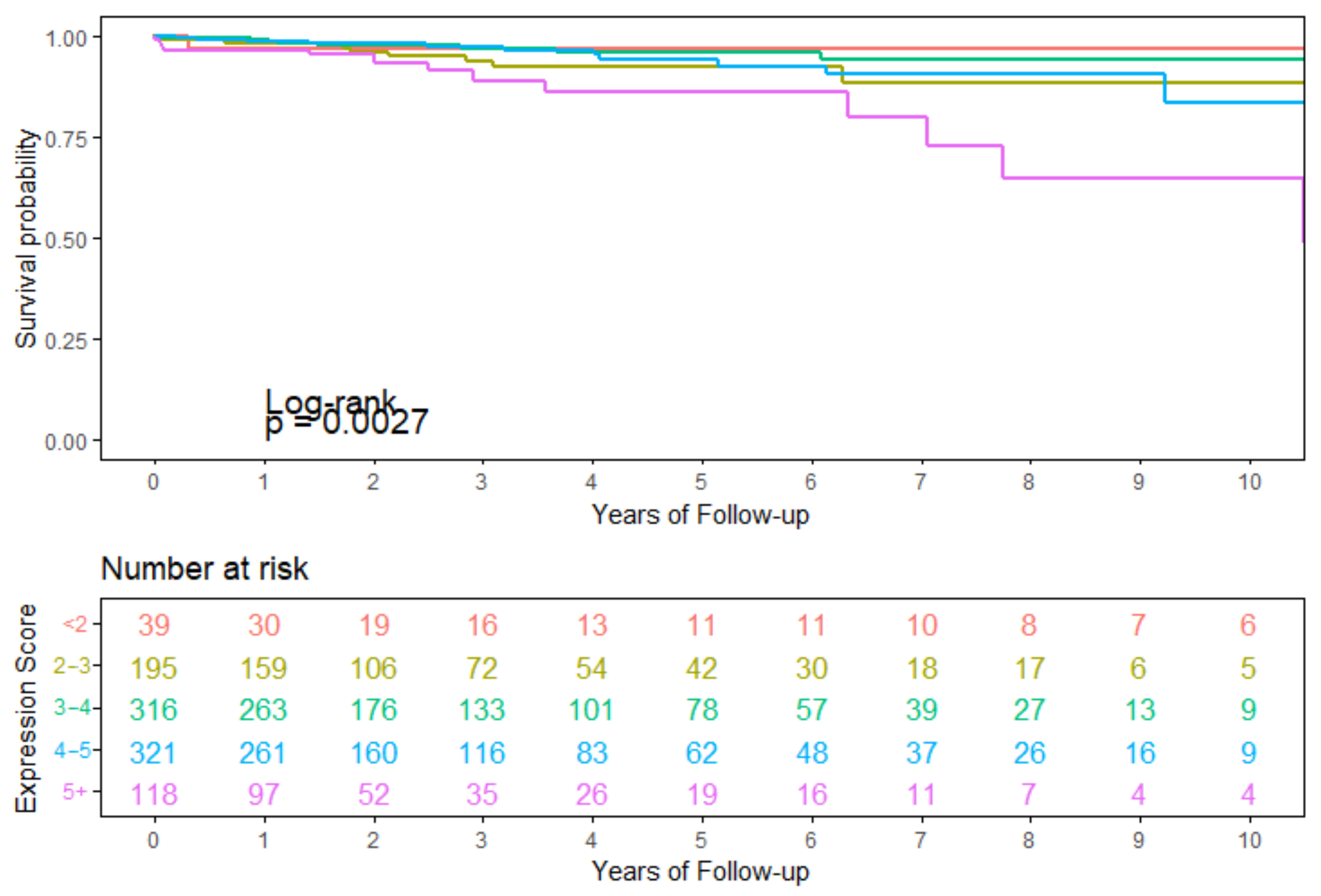
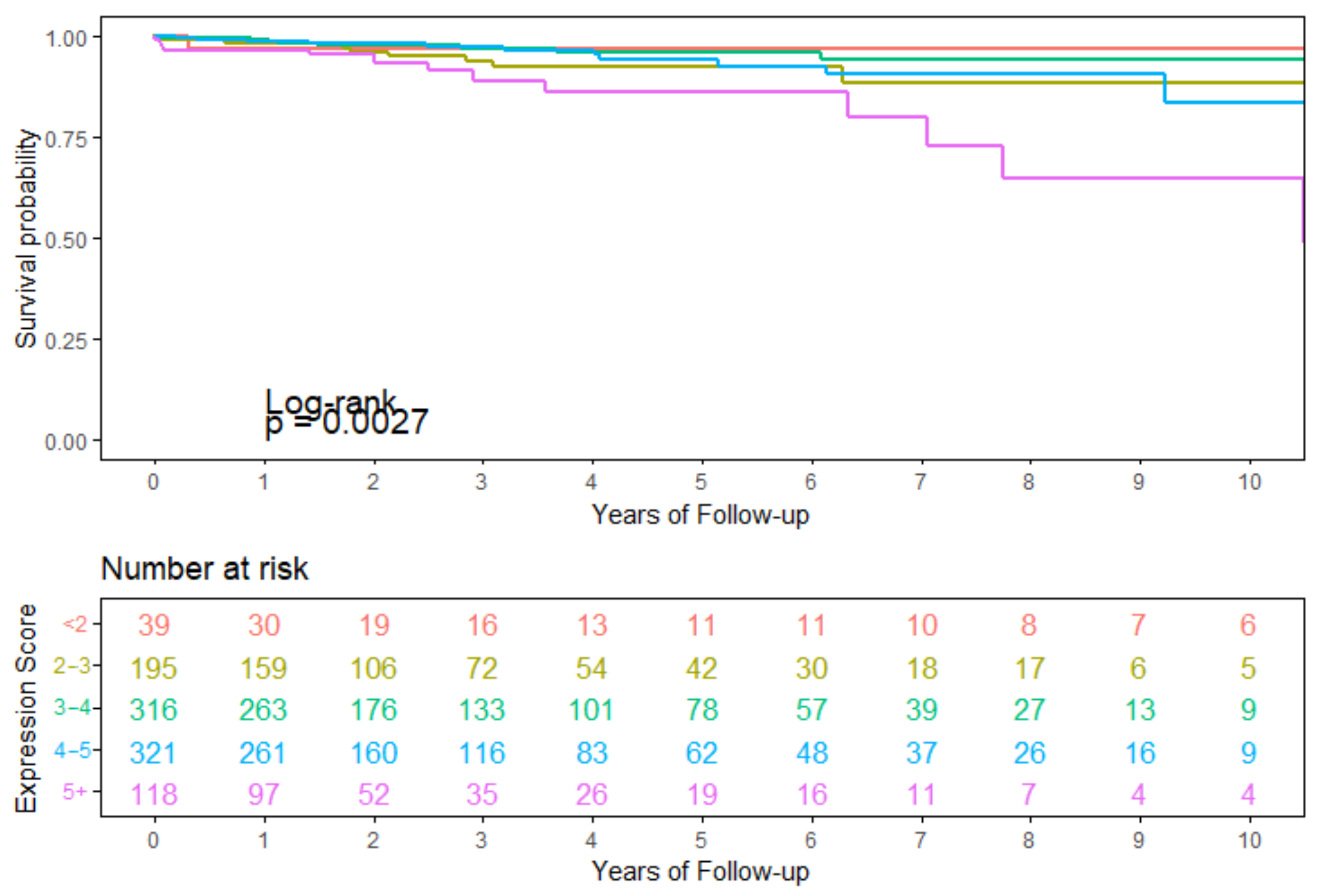
3.4. Survival Analysis Using Seven Overlapping Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GSEA | Gene Set Enrichment Analysis |
| FDR | False Discovery Rate |
| SMOTE | Synthetic Minority Over Sampling Technique |
| ANOVA | Analysis of Variance |
| KNN | K-Nearest Neighbor |
| TCGA | The Cancer Genome Atlas |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Trials | Accuracy | Precision | Recall | F1 Score | Cohen Kappa | RUC |
|---|---|---|---|---|---|---|---|
| All markers | 1 | 0.02694 | 0 | 0 | 0 | 0 | 0.5 |
| 2 | 0.97306 | 0.97306 | 1 | 0.98635 | 0 | 0.5 | |
| 3 | 0.02694 | 0 | 0 | 0 | 0 | 0.5 | |
| 4 | 0.97306 | 0.97306 | 1 | 0.98635 | 0 | 0.5 | |
| 5 | 0.97306 | 0.97306 | 1 | 0.98635 | 0 | 0.5 | |
| Avg. | 0.59461 | 0.58384 | 0.6 | 0.59181 | 0 | 0.5 | |
| St.dev. | 0.51822 | 0.53297 | 0.54772 | 0.54025 | 0 | 0 | |
| Anova_RF | 1 | 0.50168 | 1 | 0.48789 | 0.65581 | 0.04882 | 0.74395 |
| 2 | 0.92593 | 0.99628 | 0.92734 | 0.96057 | 0.36216 | 0.90117 | |
| 3 | 0.52525 | 1 | 0.51211 | 0.67735 | 0.05352 | 0.75606 | |
| 4 | 0.49158 | 1 | 0.47751 | 0.64637 | 0.04692 | 0.73875 | |
| 5 | 0.63636 | 1 | 0.62630 | 0.77021 | 0.08281 | 0.81315 | |
| Avg. | 0.61616 | 0.99926 | 0.60623 | 0.74206 | 0.11885 | 0.79061 | |
| St.dev. | 0.18252 | 0.00166 | 0.18904 | 0.13165 | 0.13679 | 0.06854 | |
| Smote | 1 | 0.98370 | 0.96842 | 1 | 0.98396 | 0.96739 | 0.98370 |
| 2 | 0.99457 | 1 | 0.98913 | 0.99454 | 0.98913 | 0.99457 | |
| 3 | 0.97826 | 1 | 0.95652 | 0.97778 | 0.95652 | 0.97826 | |
| 4 | 0.97283 | 0.97802 | 0.96739 | 0.97268 | 0.94565 | 0.97283 | |
| 5 | 0.97283 | 0.98876 | 0.95652 | 0.97238 | 0.94565 | 0.97283 | |
| Avg. | 0.98043 | 0.98704 | 0.97391 | 0.98027 | 0.96087 | 0.98043 | |
| St.dev. | 0.00909 | 0.01385 | 0.01975 | 0.00926 | 0.01819 | 0.00909 |
| Dataset | Trials | Accuracy | Precision | Recall | F1 Score | Cohen Kappa | ROC AUC |
|---|---|---|---|---|---|---|---|
| All markers base model | 1 | 0.04231 | 0 | 0 | 0 | 0 | 0.5 |
| 2 | 0.95769 | 0.95769 | 1 | 0.97839 | 0 | 0.5 | |
| 3 | 0.95769 | 0.95769 | 1 | 0.97839 | 0 | 0.5 | |
| 4 | 0.04231 | 0 | 0 | 0 | 0 | 0.5 | |
| 5 | 0.95769 | 0.95769 | 1 | 0.97839 | 0 | 0.5 | |
| Avg. | 0.59154 | 0.57461 | 0.6 | 0.58703 | 0 | 0.5 | |
| St.dev. | 0.50137 | 0.52455 | 0.54772 | 0.53588 | 0 | 0 | |
| AnovaRF base model | 1 | 0.79831 | 0.99814 | 0.79087 | 0.8825 | 0.23336 | 0.87877 |
| 2 | 0.72355 | 0.99589 | 0.71429 | 0.8319 | 0.15957 | 0.82381 | |
| 3 | 0.72496 | 1 | 0.71281 | 0.83233 | 0.17359 | 0.85641 | |
| 4 | 0.83216 | 0.99822 | 0.82622 | 0.90411 | 0.27686 | 0.89644 | |
| 5 | 0.79408 | 1 | 0.78498 | 0.87954 | 0.23603 | 0.89249 | |
| Avg. | 0.77461 | 0.99845 | 0.76583 | 0.86608 | 0.21588 | 0.86958 | |
| St.dev. | 0.04828 | 0.00169 | 0.05027 | 0.03242 | 0.04845 | 0.03 | |
| AnovaRF- SMOTE base model | 1 | 0.98889 | 1 | 0.97778 | 0.98876 | 0.97778 | 0.98889 |
| 2 | 0.97778 | 1 | 0.95556 | 0.97727 | 0.95556 | 0.97778 | |
| 3 | 0.96889 | 1 | 0.93778 | 0.96789 | 0.93778 | 0.96889 | |
| 4 | 0.97778 | 1 | 0.95556 | 0.97727 | 0.95556 | 0.97778 | |
| 5 | 0.96222 | 0.99524 | 0.92889 | 0.96092 | 0.92444 | 0.96222 | |
| Avg. | 0.97511 | 0.99905 | 0.95111 | 0.97442 | 0.95022 | 0.97511 | |
| St.dev. | 0.01011 | 0.00213 | 0.01886 | 0.01057 | 0.02022 | 0.01011 |
| Dataset | Trials | Accuracy | Precision | Recall | F1 Score | Cohen Kappa | ROC AUC |
|---|---|---|---|---|---|---|---|
| All markers large model | 1 | 0.95769 | 0.95769 | 1 | 0.97839 | 0 | 0.5 |
| 2 | 0.04231 | 0 | 0 | 0 | 0 | 0.5 | |
| 3 | 0.04231 | 0 | 0 | 0 | 0 | 0.5 | |
| 4 | 0.95769 | 0.95769 | 1 | 0.97839 | 0 | 0.5 | |
| 5 | 0.04231 | 0 | 0 | 0 | 0 | 0.5 | |
| Avg. | 0.40846 | 0.38307 | 0.4 | 0.39135 | 0 | 0.5 | |
| St.dev. | 0.50137 | 0.52455 | 0.54772 | 0.53588 | 0 | 0 | |
| AnovaRF large model | 1 | 0.93089 | 0.99685 | 0.93078 | 0.96268 | 0.50331 | 0.93206 |
| 2 | 0.89563 | 0.99672 | 0.89396 | 0.94255 | 0.39113 | 0.91365 | |
| 3 | 0.86601 | 1 | 0.86009 | 0.92478 | 0.3422 | 0.93004 | |
| 4 | 0.95628 | 1 | 0.95435 | 0.97664 | 0.63885 | 0.97717 | |
| 5 | 0.91537 | 0.99839 | 0.91311 | 0.95385 | 0.45727 | 0.93989 | |
| Avg. | 0.91283 | 0.99839 | 0.91046 | 0.9521 | 0.46656 | 0.93856 | |
| St.dev. | 0.03431 | 0.00161 | 0.0359 | 0.01971 | 0.11432 | 0.0236 | |
| AnovaRF-SMOTE large model | 1 | 0.98222 | 0.99543 | 0.96889 | 0.98198 | 0.96444 | 0.98222 |
| 2 | 0.99556 | 1 | 0.99111 | 0.99554 | 0.99111 | 0.99556 | |
| 3 | 0.98667 | 1 | 0.97333 | 0.98649 | 0.97333 | 0.98667 | |
| 4 | 0.98 | 1 | 0.96 | 0.97959 | 0.96 | 0.98 | |
| 5 | 0.99333 | 1 | 0.98667 | 0.99329 | 0.98667 | 0.99333 | |
| Avg. | 0.98756 | 0.99909 | 0.976 | 0.98738 | 0.97511 | 0.98756 | |
| St.dev. | 0.00678 | 0.00204 | 0.0128 | 0.00693 | 0.01355 | 0.00678 |
| 27K | Original | AnovaRF | SMOTE | ||||
| Normal | Cancer | Normal | Cancer | Normal | Cancer | ||
| Normal | 3.2 | 4.8 | 7.8 | 0.2 | 90.8 | 1.2 | |
| Cancer | 115.6 | 173.4 | 113.8 | 175.2 | 2.4 | 89.6 | |
| Prediction Sample Size | 297 | 297 | 184 | ||||
| 450K base model | Original | AnovaRF | SMOTE | ||||
| Normal | Cancer | Normal | Cancer | Normal | Cancer | ||
| Normal | 12 | 18 | 28.2 | 1.8 | 224.8 | 0.2 | |
| Cancer | 271.6 | 407.4 | 150 | 529 | 11 | 214 | |
| Prediction Sample Size | 709 | 709 | 450 | ||||
| 450K larger model | Original | AnovaRF | SMOTE | ||||
| Normal | Cancer | Normal | Cancer | Normal | Cancer | ||
| Normal | 18 | 12 | 28.4 | 1.6 | 224.8 | 0.2 | |
| Cancer | 407.4 | 271.6 | 86 | 593 | 5.4 | 219.6 | |
| Prediction Sample Size | 709 | 709 | 450 | ||||
Appendix B
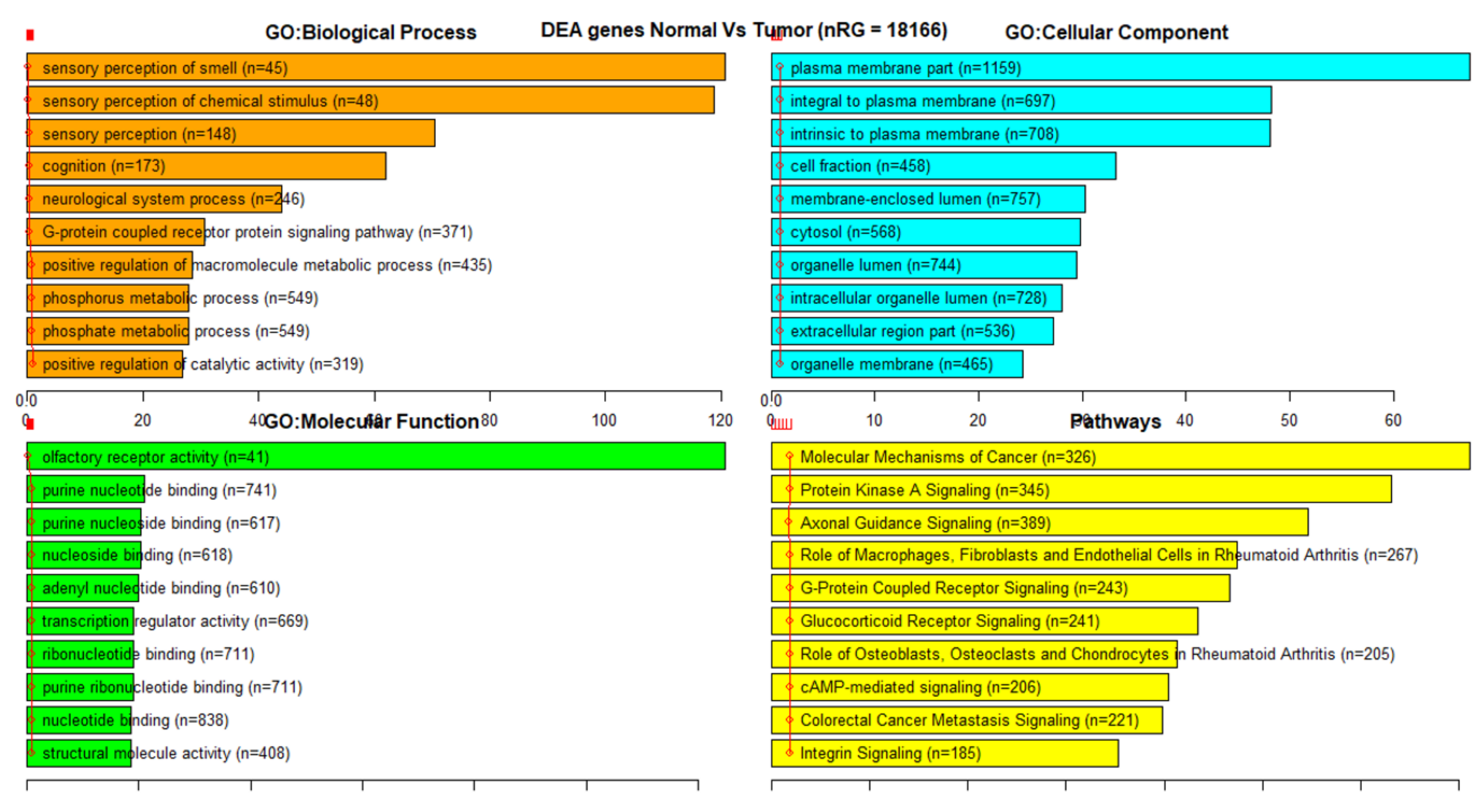

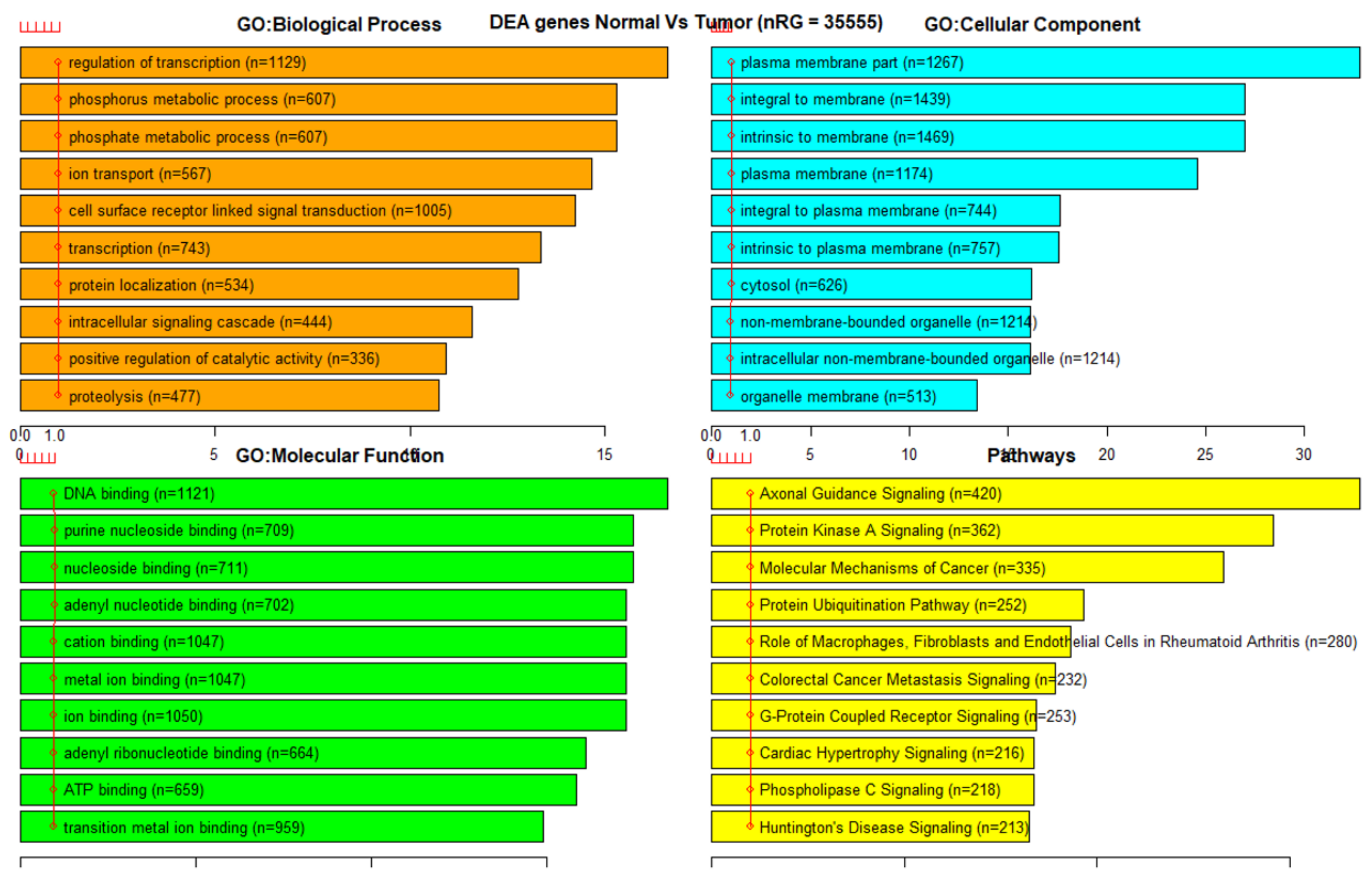
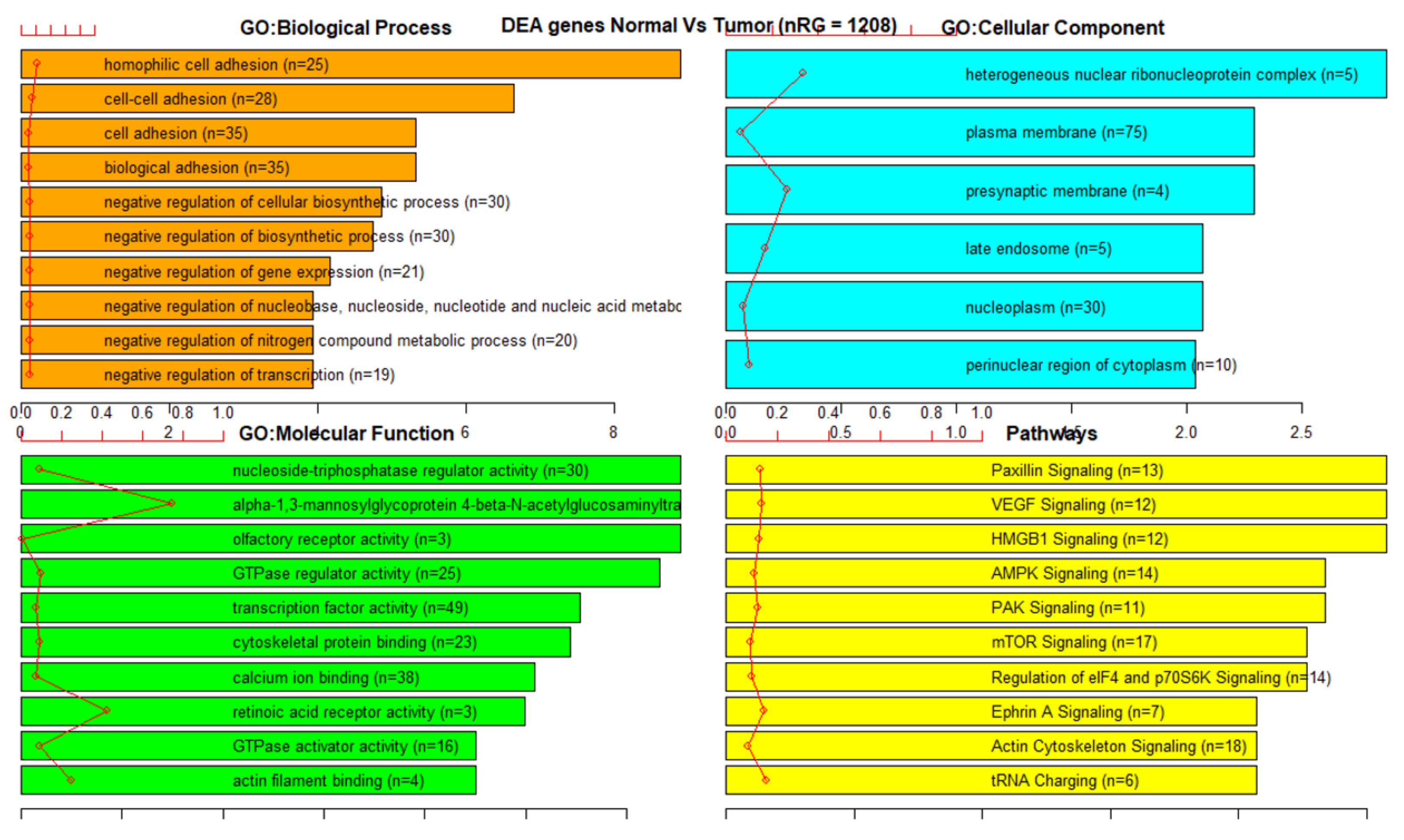
References
- Xiao, C.L.; Zhu, S.; He, M.; Chen, D.; Zhang, Q.; Chen, Y.; Yu, G.; Liu, J.; Xie, S.Q.; Luo, F.; et al. N6-methyladenine DNA modification in the human genome. Mol. Cell 2018, 71, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Levin, J.Z.; Yassour, M.; Adiconis, X.; Nusbaum, C.; Thompson, D.A.; Friedman, N.; Gnirke, A.; Regev, A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 2010, 7, 709–715. [Google Scholar] [CrossRef] [PubMed]
- IlluminaHumanMethylation450kmanifest: Annotation for Illumina’s 450k Methylation Arrays. Available online: https://bioconductor.org/packages/release/data/annotation/html/IlluminaHumanMethylation450kmanifest.html (accessed on 10 August 2022).
- O’Shea, K.; Nash, R. An introduction to convolutional neural networks. arXiv 2015, arXiv:1511.08458. [Google Scholar]
- Zaremba, W.; Sutskever, I.; Vinyals, O. Recurrent neural network regularization. arXiv 2014, arXiv:1409.2329. [Google Scholar]
- Halevy, A.; Norvig, P.; Pereira, F. The unreasonable effectiveness of data. IEEE Intell. Syst. 2009, 24, 8–12. [Google Scholar] [CrossRef]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. Imagenet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 2012, 25. Available online: https://proceedings.neurips.cc/paper/2012/hash/c399862d3b9d6b76c8436e924a68c45b-Abstract.html (accessed on 10 August 2022). [CrossRef]
- Johnson, R.; Zhang, T. Effective use of word order for text categorization with convolutional neural networks. arXiv 2014, arXiv:1412.1058. [Google Scholar]
- Verleysen, M.; François, D. The curse of dimensionality in data mining and time series prediction. In International Work-Conference on Artificial Neural Networks; Springer: Berlin/Heidelberg, Germany, 2005; pp. 758–770. [Google Scholar]
- Ahsan, M.; Gomes, R.; Chowdhury, M.; Nygard, K.E. Enhancing Machine Learning Prediction in Cybersecurity Using Dynamic Feature Selector. J. Cybersecur. Priv. 2021, 1, 199–218. [Google Scholar] [CrossRef]
- Longadge, R.; Dongre, S. Class imbalance problem in data mining review. arXiv 2013, arXiv:1305.1707. [Google Scholar]
- Wang, Y.; Liu, T.; Xu, D.; Shi, H.; Zhang, C.; Mo, Y.Y.; Wang, Z. Predicting DNA methylation state of CpG dinucleotide using genome topological features and deep networks. Sci. Rep. 2016, 6, 19598. [Google Scholar] [CrossRef]
- Angermueller, C.; Lee, H.J.; Reik, W.; Stegle, O. DeepCpG: Accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 2017, 18, 67. [Google Scholar]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016, 26, 304–319. [Google Scholar] [CrossRef]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef]
- Ni, P.; Huang, N.; Zhang, Z.; Wang, D.P.; Liang, F.; Miao, Y.; Xiao, C.L.; Luo, F.; Wang, J. DeepSignal: Detecting DNA methylation state from Nanopore sequencing reads using deep-learning. Bioinformatics 2019, 35, 4586–4595. [Google Scholar] [CrossRef]
- Liu, B.; Liu, Y.; Pan, X.; Li, M.; Yang, S.; Li, S.C. DNA methylation markers for pan-cancer prediction by deep learning. Genes 2019, 10, 778. [Google Scholar] [CrossRef]
- Tian, Q.; Zou, J.; Tang, J.; Fang, Y.; Yu, Z.; Fan, S. MRCNN: A deep learning model for regression of genome-wide DNA methylation. BMC Genom. 2019, 20, 192. [Google Scholar] [CrossRef]
- Heath, A.P.; Ferretti, V.; Agrawal, S.; An, M.; Angelakos, J.C.; Arya, R.; Bajari, R.; Baqar, B.; Barnowski, J.H.; Burt, J.; et al. The NCI genomic data commons. Nat. Genet. 2021, 53, 257–262. [Google Scholar] [CrossRef]
- Di Lena, P.; Sala, C.; Prodi, A.; Nardini, C. Missing value estimation methods for DNA methylation data. Bioinformatics 2019, 35, 3786–3793. [Google Scholar] [CrossRef]
- Troyanskaya, O.; Cantor, M.; Sherlock, G.; Brown, P.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing value estimation methods for DNA microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef]
- Tipping, M.E. Sparse Bayesian learning and the relevance vector machine. J. Mach. Learn. Res. 2001, 1, 211–244. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Kim, T.K. Understanding one-way ANOVA using conceptual figures. Korean J. Anesthesiol. 2017, 70, 22. [Google Scholar] [CrossRef]
- Gomes, R.; Ahsan, M.; Denton, A. Random forest classifier in SDN framework for user-based indoor localization. In Proceedings of the 2018 IEEE International Conference on Electro/Information Technology (EIT), Rochester, MI, USA, 3–5 May 2018; pp. 0537–0542. [Google Scholar]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. {TensorFlow}: A System for {Large-Scale} Machine Learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI 16), Savannah, GA, USA, 2–4 November 2016; pp. 265–283. [Google Scholar]
- Xu, B.; Wang, N.; Chen, T.; Li, M. Empirical evaluation of rectified activations in convolutional network. arXiv 2015, arXiv:1505.00853. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Ein-Dor, L.; Kela, I.; Getz, G.; Givol, D.; Domany, E. Outcome signature genes in breast cancer: Is there a unique set? Bioinformatics 2005, 21, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Silva, T.C.; Colaprico, A.; Olsen, C.; D’Angelo, F.; Bontempi, G.; Ceccarelli, M.; Noushmehr, H. TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Research 2016, 5, 1542. [Google Scholar] [CrossRef]
- Mounir, M.; Lucchetta, M.; Silva, T.C.; Olsen, C.; Bontempi, G.; Chen, X.; Noushmehr, H.; Colaprico, A.; Papaleo, E. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput. Biol. 2019, 15, e1006701. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.; Clements, J.; Dawson, E.; Bamford, S.; Webb, T.; Dogan, A.; Flanagan, A.; Teague, J.; Wooster, R.; Futreal, P.; et al. COSMIC 2005. Br. J. Cancer 2006, 94, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Sun, J.; Zhao, Z. TSGene: A web resource for tumor suppressor genes. Nucleic Acids Res. 2013, 41, D970–D976. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Kim, P.; Mitra, R.; Zhao, J.; Zhao, Z. TSGene 2.0: An updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2016, 44, D1023–D1031. [Google Scholar] [CrossRef]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Liang, Z.Z.; Zhang, Y.X.; Zhu, R.M.; Li, Y.L.; Jiang, H.M.; Li, R.B.; Chen, Q.X.; Wang, Q.; Tang, L.Y.; Ren, Z.F. Identification of epigenetic modifications mediating the antagonistic effect of selenium against cadmium-induced breast carcinogenesis. Environ. Sci. Pollut. Res. 2022, 29, 22056–22068. [Google Scholar] [CrossRef]
- Kominsky, S.L.; Argani, P.; Korz, D.; Evron, E.; Raman, V.; Garrett, E.; Rein, A.; Sauter, G.; Kallioniemi, O.P.; Sukumar, S. Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene 2003, 22, 2021–2033. [Google Scholar] [CrossRef] [Green Version]
- Savci-Heijink, C.; Halfwerk, H.; Koster, J.; Horlings, H.; Van De Vijver, M. A specific gene expression signature for visceral organ metastasis in breast cancer. BMC Cancer 2019, 19, 333. [Google Scholar] [CrossRef]
- Koo, J.; Cabarcas-Petroski, S.; Petrie, J.L.; Diette, N.; White, R.J.; Schramm, L. Induction of proto-oncogene BRF2 in breast cancer cells by the dietary soybean isoflavone daidzein. BMC Cancer 2015, 15, 905. [Google Scholar] [CrossRef]
- Di Emidio, G.; D’Aurora, M.; Placidi, M.; Franchi, S.; Rossi, G.; Stuppia, L.; Artini, P.G.; Tatone, C.; Gatta, V. Pre-conceptional maternal exposure to cyclophosphamide results in modifications of DNA methylation in F1 and F2 mouse oocytes: Evidence for transgenerational effects. Epigenetics 2019, 14, 1057–1064. [Google Scholar] [CrossRef]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [Green Version]



| Dataset | Total Samples | Tumor Samples | Normal Samples | # CpG Markers |
|---|---|---|---|---|
| 27K | 337 | 309 | 28 | 27,578 |
| 450K | 851 | 750 | 101 | 485,577 |
| Metric | Zero Impute | KNN Impute | Mean Impute | Iterative Impute | |
|---|---|---|---|---|---|
| 27K | MSE | 0.016648 | 0.016755 | 0.016749 | 0.016777 |
| STD | 0.007299 | 0.007253 | 0.007245 | 0.007340 | |
| 450K | MSE | 0.017244 | 0.017253 | 0.017251 | ——– |
| STD | 0.005273 | 0.005286 | 0.005307 | ——– |
| Dataset | # Features | Sample Size | Tumor Samples | Normal Samples | Runtime | |
|---|---|---|---|---|---|---|
| 27K | All markers | 24,981 | 337 | 309 | 28 | 21 s |
| Anova_RF | 336 | 337 | 309 | 28 | 12 s | |
| Anova_RF (with Smote) | 475 | 618 | 309 | 309 | 13 s | |
| 450K | 450K All (base + large) | 395,722 | 851 | 750 | 101 | 1:44:10 s |
| Anova_RF (base + large) | 1044 | 851 | 750 | 101 | 38:41 s | |
| Anova_RF with SMOTE (base + large) | 1445 | 1500 | 525 | 525 | 13 s |
| Dataset | CpG Markers | Total Genes | COSMIC + TSGene Overlap (3326 Genes) | Sample Genes Overlap (100 Genes) |
|---|---|---|---|---|
| 27K all | 24,981 | 18,166 | 1214 | 98 |
| 27K ANOVA-RF | 336 | 470 | 36 | 2 |
| 27K ANOVA-RF SMOTE | 475 | 685 | 55 | 6 |
| 450K all | 395,722 | 35,555 | 1455 | 100 |
| 450K ANOVA-RF | 1044 | 1208 | 88 | 7 |
| 450K ANOVA-RF SMOTE | 1445 | 1572 | 136 | 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, R.; Paul, N.; He, N.; Huber, A.F.; Jansen, R.J. Application of Feature Selection and Deep Learning for Cancer Prediction Using DNA Methylation Markers. Genes 2022, 13, 1557. https://doi.org/10.3390/genes13091557
Gomes R, Paul N, He N, Huber AF, Jansen RJ. Application of Feature Selection and Deep Learning for Cancer Prediction Using DNA Methylation Markers. Genes. 2022; 13(9):1557. https://doi.org/10.3390/genes13091557
Chicago/Turabian StyleGomes, Rahul, Nijhum Paul, Nichol He, Aaron Francis Huber, and Rick J. Jansen. 2022. "Application of Feature Selection and Deep Learning for Cancer Prediction Using DNA Methylation Markers" Genes 13, no. 9: 1557. https://doi.org/10.3390/genes13091557
APA StyleGomes, R., Paul, N., He, N., Huber, A. F., & Jansen, R. J. (2022). Application of Feature Selection and Deep Learning for Cancer Prediction Using DNA Methylation Markers. Genes, 13(9), 1557. https://doi.org/10.3390/genes13091557