
Inherited Retinal Dystrophy in Southeastern United States: Characterization of South Carolina Patients and Comparative Literature Review

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Demographics
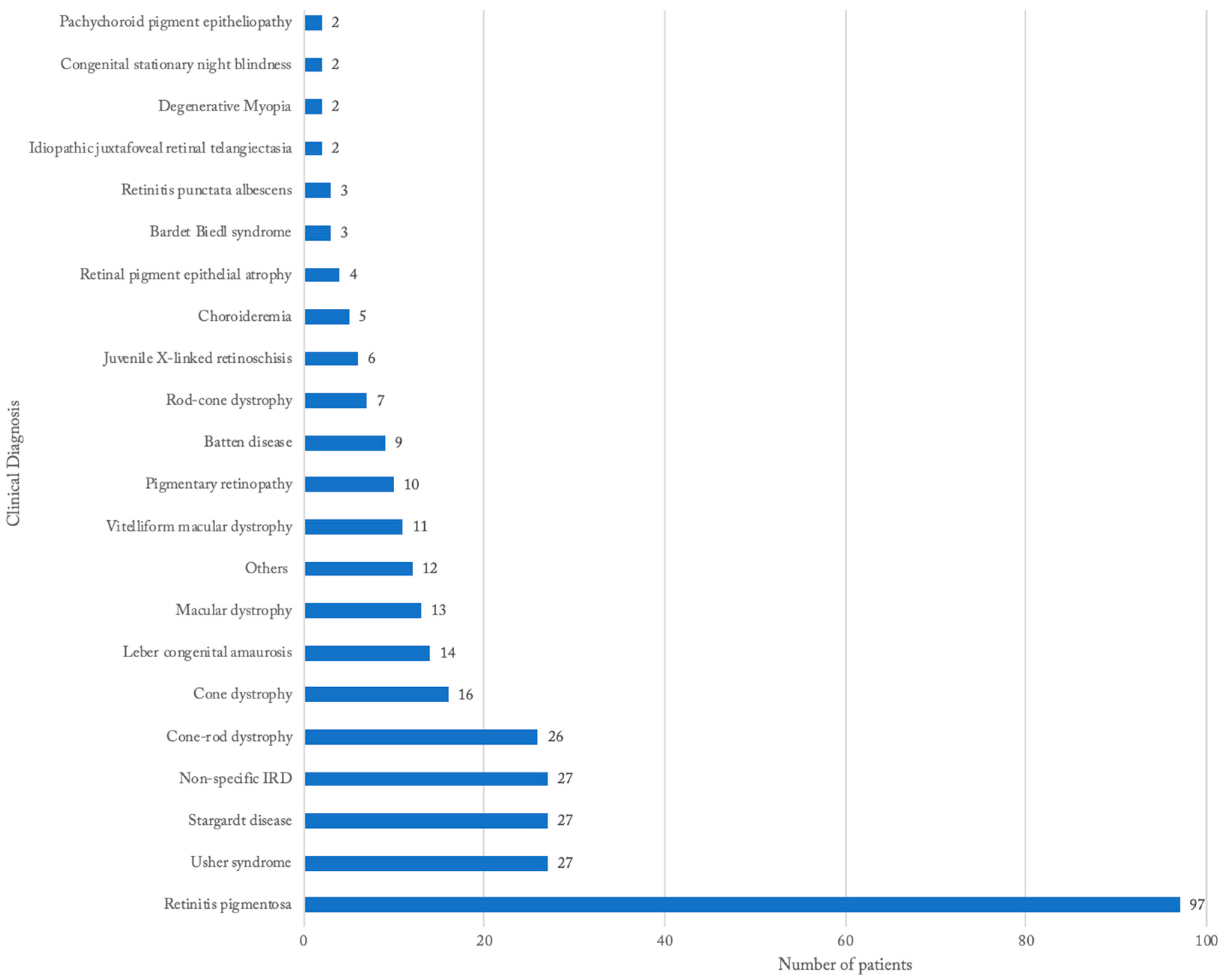
3.2. Distribution of IRDs Based on Clinical Diagnosis
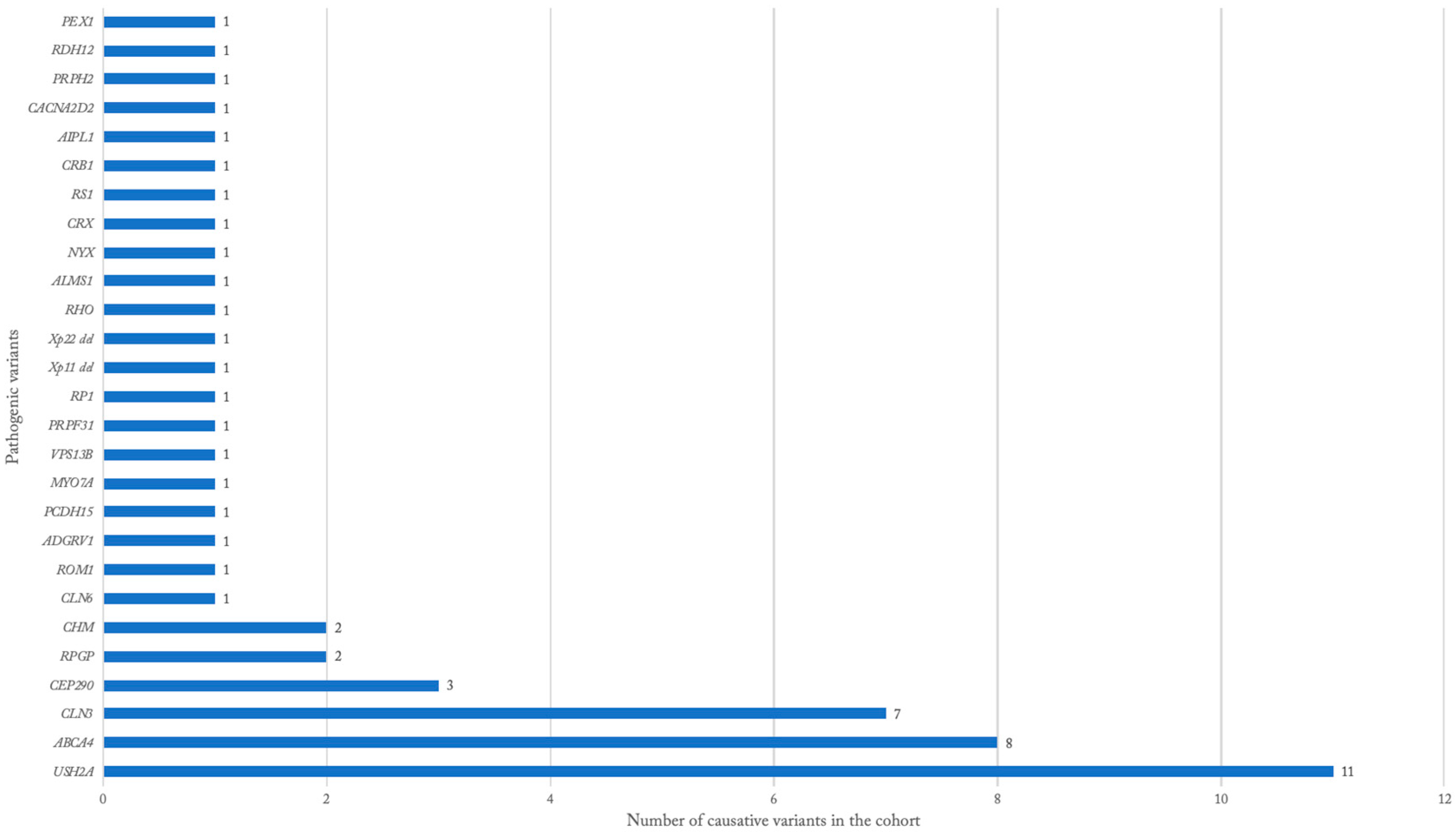
3.3. Genetic Findings
3.4. Disease Characteristics
3.5. National and International Comparisons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Solebo, A.L.; Teoh, L.; Rahi, J. Epidemiology of blindness in children. Arch. Dis. Child 2017, 102, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C.P. Gene discovery and prevalence in inherited retinal dystrophies. C. R. Biol. 2014, 337, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Sitorus, R.S.; Abidin, M.S.; Prihartono, J. Causes and temporal trends of childhood blindness in Indonesia: Study at schools for the blind in Java. Br. J. Ophthalmol. 2007, 91, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Bhargava, A.; George, R.; Ve Ramesh, S.; Hemamalini, A.; Prema, R.; Kumaramanickavel, G.; Vijaya, L. Prevalence of retinitis pigmentosa in South Indian population aged above 40 years. Ophthalmic Epidemiol. 2008, 15, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.I.; Muecke, J.; Hammerton, M.; Ngy, M.; Kong, A.; Morse, A.; Holmes, M.; Piseth, H.; Hamilton, C.; Selva, D. A survey of visual impairment and blindness in children attending four schools for the blind in Cambodia. Ophthalmic Epidemiol. 2010, 17, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Sohocki, M.M.; Daiger, S.P.; Bowne, S.J.; Rodriquez, J.A.; Northrup, H.; Heckenlively, J.R.; Birch, D.G.; Mintz-Hittner, H.; Ruiz, R.S.; Lewis, R.A.; et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 2001, 17, 42–51. [Google Scholar] [CrossRef]
- Puech, B.; Kostrubiec, B.; Hache, J.C.; François, P. Epidemiology and prevalence of hereditary retinal dystrophies in the Northern France. J. Fr. Ophtalmol. 1991, 14, 153–164. [Google Scholar]
- Bertelsen, M.; Jensen, H.; Bregnhøj, J.F.; Rosenberg, T. Prevalence of generalized retinal dystrophy in Denmark. Ophthalmic Epidemiol. 2014, 21, 217–223. [Google Scholar] [CrossRef]
- Bocquet, B.; Lacroux, A.; Surget, M.O.; Baudoin, C.; Marquette, V.; Manes, G.; Hebrard, M.; Sénéchal, A.; Delettre, C.; Roux, A.F.; et al. Relative frequencies of inherited retinal dystrophies and optic neuropathies in Southern France: Assessment of 21-year data management. Ophthalmic Epidemiol. 2013, 20, 13–25. [Google Scholar] [CrossRef]
- Al-Merjan, J.I.; Pandova, M.G.; Al-Ghanim, M.; Al-Wayel, A.; Al-Mutairi, S. Registered blindness and low vision in Kuwait. Ophthalmic Epidemiol. 2005, 12, 251–257. [Google Scholar] [CrossRef]
- Xu, L.; Hu, L.; Ma, K.; Li, J.; Jonas, J.B. Prevalence of retinitis pigmentosa in urban and rural adult Chinese: The Beijing Eye Study. Eur. J. Ophthalmol. 2006, 16, 865–866. [Google Scholar] [CrossRef] [PubMed]
- Tous, H.M.; Izquierdo, N.J. Retinitis pigmentosa in Puerto Rico. P. R. Health Sci. J. 2006, 25, 315–318. [Google Scholar] [PubMed]
- Boughman, J.A.; Vernon, M.; Shaver, K.A. Usher syndrome: Definition and estimate of prevalence from two high-risk populations. J. Chronic Dis. 1983, 36, 595–603. [Google Scholar] [CrossRef]
- Blacharski, P. Fundus flavimaculatus. In Retinal Dystrophies and Degenerations; Newsome, D.A., Ed.; Raven Press: New York, NY, USA, 1988. [Google Scholar]
- Allikmets, R. Leber congenital amaurosis: A genetic paradigm. Ophthalmic Genet. 2004, 25, 67–79. [Google Scholar] [CrossRef]
- Nordström, S. Hereditary macular degeneration—A population survey in the country of Vsterbotten, Sweden. Hereditas 1974, 78, 41–62. [Google Scholar] [CrossRef]
- George, N.D.; Yates, J.R.; Moore, A.T. X linked retinoschisis. Br. J. Ophthalmol. 1995, 79, 697–702. [Google Scholar] [CrossRef]
- George, N.D.; Yates, J.R.; Moore, A.T. Clinical features in affected males with X-linked retinoschisis. Arch. Ophthalmol. 1996, 114, 274–280. [Google Scholar] [CrossRef]
- Daiger, S. Summaries of Genes and Loci Causing Retinal Diseases; The University of Texas Health Science Center: Houston, TX, USA, 2021. [Google Scholar]
- Goetz, K.E.; Reeves, M.J.; Gagadam, S.; Blain, D.; Bender, C.; Lwin, C.; Naik, A.; Tumminia, S.J.; Hufnagel, R.B. Genetic testing for inherited eye conditions in over 6000 individuals through the eyeGENE network. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 828–837. [Google Scholar] [CrossRef]
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef]
- Perea-Romero, I.; Gordo, G.; Iancu, I.F.; Del Pozo-Valero, M.; Almoguera, B.; Blanco-Kelly, F.; Carreño, E.; Jimenez-Rolando, B.; Lopez-Rodriguez, R.; Lorda-Sanchez, I.; et al. Author Correction: Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci. Rep. 2021, 11, 10340. [Google Scholar] [CrossRef]
- Colombo, L.; Maltese, P.E.; Castori, M.; El Shamieh, S.; Zeitz, C.; Audo, I.; Zulian, A.; Marinelli, C.; Benedetti, S.; Costantini, A.; et al. Molecular Epidemiology in 591 Italian Probands With Nonsyndromic Retinitis Pigmentosa and Usher Syndrome. Investig. Ophthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar] [CrossRef] [PubMed]
- Motta, F.L.; Martin, R.P.; Filippelli-Silva, R.; Salles, M.V.; Sallum, J.M.F. Relative frequency of inherited retinal dystrophies in Brazil. Sci. Rep. 2018, 8, 15939. [Google Scholar] [CrossRef] [PubMed]
- Whelan, L.; Dockery, A.; Wynne, N.; Zhu, J.; Stephenson, K.; Silvestri, G.; Turner, J.; O’Byrne, J.J.; Carrigan, M.; Humphries, P.; et al. Findings from a Genotyping Study of Over 1000 People with Inherited Retinal Disorders in Ireland. Genes 2020, 11, 105. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Ben-Yosef, T.; Goldenberg-Cohen, N.; Pras, E.; Gradstein, L.; Soudry, S.; Mezer, E.; Zur, D.; Abbasi, A.H.; Zeitz, C.; et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 2020, 41, 140–149. [Google Scholar] [CrossRef] [PubMed]
- 2020 Census Demographic Data Map Viewer. Available online: https://www.census.gov/library/visualizations/2021/geo/demographicmapviewer.html (accessed on 15 January 2022).
- Pontikos, N.; Arno, G.; Jurkute, N.; Schiff, E.; Ba-Abbad, R.; Malka, S.; Gimenez, A.; Georgiou, M.; Wright, G.; Armengol, M.; et al. Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More Than 3000 Families from the United Kingdom. Ophthalmology 2020, 127, 1384–1394. [Google Scholar] [CrossRef]
- Weisschuh, N.; Obermaier, C.D.; Battke, F.; Bernd, A.; Kuehlewein, L.; Nasser, F.; Zobor, D.; Zrenner, E.; Weber, E.; Wissinger, B.; et al. Genetic architecture of inherited retinal degeneration in Germany: A large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 2020, 41, 1514–1527. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Chen, N.; Zhang, F.; Ma, Z.; Li, G.; Yang, L. Application of Whole Exome and Targeted Panel Sequencing in the Clinical Molecular Diagnosis of 319 Chinese Families with Inherited Retinal Dystrophy and Comparison Study. Genes 2018, 9, 360. [Google Scholar] [CrossRef]
- Kimberling, W.J.; Hildebrand, M.S.; Shearer, A.E.; Jensen, M.L.; Halder, J.A.; Trzupek, K.; Cohn, E.S.; Weleber, R.G.; Stone, E.M.; Smith, R.J. Frequency of Usher syndrome in two pediatric populations: Implications for genetic screening of deaf and hard of hearing children. Genet. Med. 2010, 12, 512–516. [Google Scholar] [CrossRef]
- Hamel, C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef]
- Dalvin, L.A.; Pulido, J.S.; Marmorstein, A.D. Vitelliform dystrophies: Prevalence in Olmsted County, Minnesota, United States. Ophthalmic Genet. 2017, 38, 143–147. [Google Scholar] [CrossRef]
- Bitner, H.; Schatz, P.; Mizrahi-Meissonnier, L.; Sharon, D.; Rosenberg, T. Frequency, genotype, and clinical spectrum of best vitelliform macular dystrophy: Data from a national center in Denmark. Am. J. Ophthalmol. 2012, 154, 403–412.e4. [Google Scholar] [CrossRef] [PubMed]
- Batten Disease Fact Sheet; National Institutes of Health and National Institute of Neurological Disorders and Stroke: Bethesda, MD, USA, 2018.
- Khan, K.N.; Islam, F.; Moore, A.T.; Michaelides, M. Clinical and Genetic Features of Choroideremia in Childhood. Ophthalmology 2016, 123, 2158–2165. [Google Scholar] [CrossRef] [PubMed]
- Mataftsi, A.; Zografos, L.; Millá, E.; Secrétan, M.; Munier, F.L. Bietti’s crystalline corneoretinal dystrophy: A cross-sectional study. Retina 2004, 24, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Printzlau, A.; Andersen, M. Pierre Robin sequence in Denmark: A retrospective population-based epidemiological study. Cleft Palate Craniofac. J. 2004, 41, 47–52. [Google Scholar] [CrossRef]
- Christensen, D.R.G.; Brown, F.E.; Cree, A.J.; Ratnayaka, J.A.; Lotery, A.J. Sorsby fundus dystrophy—A review of pathology and disease mechanisms. Exp. Eye Res. 2017, 165, 35–46. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Confirmed Disease-Causing Genes (n) |
|---|---|
| Alstrom cone dystrophy | ALMS1 (1) |
| Batten disease | CLN3 (7), CLN6 (1) |
| Choroideremia | CHM (2) |
| Cohen syndrome | VPS13B (1) |
| Cone-rod dystrophy | RPGR (1), USH2A (1) |
| Congenital stationary night blindness | NYX (1) |
| Juvenile X-linked retinoschisis | RS1 (1) |
| Leber congenital amaurosis | CEP-290 (3), AIPL1 (1), CACNA2D2 (1), CRB1 (1) |
| Opitz G/BBB syndrome | Xp22 deletion (1) |
| Pigmentary retinopathy | PRPH2 (1) |
| Retinitis pigmentosa | PRPF31 (1), RDH12 (1), RHO (1), RP1 (1), RPGR (1), RP1L1 (1), ROM1 (1), Xp11 del (1) |
| Stargardt disease | ABCA4 (8) |
| Non-specific IRD | CRX (1) |
| Usher syndrome | USH2A (10), ADGRV1 (1), MYO7A (1), PCDH15 (1) |
| Zellweger Syndrome | PEX1 (1) |
| Disease | n | Age Range | Visual Acuity Range | Positive Family History | Race | Retinal Findings | Other Ocular Pathologies |
|---|---|---|---|---|---|---|---|
| Retinitis pigmentosa | 97 | 5 to 78 | 20/20 to NLP | 31% | White (55%), Black (44%), Hispanic (1%) | bony spicules (69%), vascular attenuation (62%), optic nerve pallor (36%), atrophy (18%), pigment mottling/clumping (15%) | early cataract (16%), nystagmus (9%), strabismus (7%), glaucoma (7%), cystoid macular edema (4%), keratoconus (2%) |
| Usher syndrome | 27 | 5 to 60 | 20/20 to HM | 27% | White (62%), Black (29%), Hispanic (5%), Asian (5%) | bony spicules (56%), vascular attenuation (44%), optic nerve pallor (31%), normal (19%), atrophy (13%) | early cataract (7%), hyperopic astigmatism (7%) vitreous detachment (4%), glaucoma (4%), diplopia (4%), corneal clouding (4%), Sjogren’s syndrome (4%) |
| Stargardt disease | 27 | 10 to 79 | 20/30 to 20/800 | 42% | White (77%), Black (23%) | macular atrophy (48%), flecks (32%), pigmentary changes (28%), bull’s eye (20%), beaten metal appearance (8%) | myopic astigmatism (8%), strabismus (4%), posterior vitreous detachment (4%), asteroid hyalosis (4%) |
| Cone-rod dystrophy | 26 | 12 to 57 | 20/20 to LP | 38% | White (60%), Black (30%), Hispanic (10%) | pigmentary changes (30%), bone spicules (22%), attenuation (22%), pallor (22%), RPE changes (13%), atrophy (13%), foveal hypopigmentation (9%), tapetal reflex (9%), normal (9%) | myopia (12%), strabismus (8%), nystagmus (8%) |
| Cone dystrophy | 16 | 8 to 74 | 20/30 to 20/800 | 38% | White (64%), Black (29%), Hispanic (7%) | atrophy (43%), bull’s eye maculopathy (21%), normal (21%), vascular attenuation (14%) | nystagmus (13%), amblyopia (6%), high myopia (6%) |
| Leber congenital amaurosis | 14 | 1 to 31 | 20/30 to NLP | 30% | White (62%), Hispanic (14%), Black (8%), Asian (8%), Bi-racial (8%) | pigmentary changes (36%), pallor (27%), attenuation (18%), atrophy (18%) | nystagmus (86), high hyperopia (43%), strabismus (36%), oculo-digital sign (14%) |
| Disease | Eye Gene Percentage | SC Percentage | Prevalence in the Literature [References] |
|---|---|---|---|
| Retinitis pigmentosa | 38.4 | 29.8 | 1 in 3000–4000 [4,7,8,9,10,11,12] |
| Usher syndrome | 3.7 | 8.3 | 1 in 6000–25,000 [13,31] |
| Stargardt disease | 24.0 | 8.3 | 1 in 8000–10,000 [14] |
| Cone-rod dystrophy * | 8.5 | 15.1 | 1 in 40,000 [32] |
| Leber congenital amaurosis | 0.9 | 4.3 | 1 in 50,000–100,000 [15] |
| Best vitelliform macular dystrophy | 4.0 | 3.4 | 1 in 16,500–21,000 [16,33,34] |
| Batten disease | 0.0 | 2.8 | 1 in 25,000–50,000 [35] |
| X-linked juvenile retinschisis | 3.3 | 1.8 | 1 in 5000–25,000 [17,18] |
| Choroideremia | 4.3 | 1.5 | 1 in 50,000–100,000 [36] |
| Doyne Honeycomb dystrophy | 1.7 | 0.3 | unknown |
| Pattern dystrophy ** | 4.9 | 0.3 | 1 in 7400–8200 [33] |
| FEVR (Familial exudative vitreoretinopathy) | 2.5 | 0 | unknown |
| Bietti crystalline corneal–retinal dystrophy | 0.5 | 0 | 1 in 100,000–135,000 [37] |
| Kearns–Sayre syndrome | 0.1 | 0 | unknown |
| Congenital stationary night blindness/Oguchi disease | 1.4 | 0.6 | unknown |
| Occult macular dystrophy | 0.6 | 0 | unknown |
| Stickler syndrome | 0.4 | 0 | 1 in 7500–9000 [38] |
| Sorsby dystrophy | 0.6 | 0 | 1 in 220,000 [39] |
| Location [References] | Number of Patients | Year | 1st Most Common Gene | 2nd Most Common Gene | 3rd Most Common Gene | 1st Most Common Disease | 2nd Most Common Disease | 3rd Most Common Disease |
|---|---|---|---|---|---|---|---|---|
| SC | 325 | 2022 | USH2A | ABCA4 | CLN3 | RP | cone-rod/rod-cone dystrophy | Usher and Stargardt tied |
| Brazil [24] | 1246 | 2018 | ABCA4 | USH2A | CEP-290 | RP | Leber congential amaurosis | Stargardt disease |
| Israel [26] | 2420 | 2020 | ABCA4 | USH2A | FAM161A | RP | Stargardt disease | cone-rod/rod-cone dystrophy |
| USA [20] | 5385 | 2020 | ABCA4 | USH2A | RPGR | RP | Stargardt disease | cone-rod/rod-cone dystrophy |
| France [9] | 1957 | 2013 | ABCA4 | USH2A | MYO7A | RP | Usher syndrome | cone-rod/rod-cone dystrophy |
| UK [28] | 4236 | 2020 | ABCA4 | USH2A | RPGR | n/a | n/a | n/a |
| Germany [29] | 2158 | 2020 | ABCA4 | USH2A | RPGR | RP | Macular Dystrophy | Cone-rod/rod-cone dystrophy |
| China [30] | 319 | 2018 | USH2A | RPGR | CYP4V2 | RP | Cone-rod/rod-cone dystrophy | Usher syndrome |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griffith, J., III; Sioufi, K.; Wilbanks, L.; Magrath, G.N.; Say, E.A.T.; Lyons, M.J.; Wilkes, M.; Pai, G.S.; Peterseim, M.M.W. Inherited Retinal Dystrophy in Southeastern United States: Characterization of South Carolina Patients and Comparative Literature Review. Genes 2022, 13, 1490. https://doi.org/10.3390/genes13081490
Griffith J III, Sioufi K, Wilbanks L, Magrath GN, Say EAT, Lyons MJ, Wilkes M, Pai GS, Peterseim MMW. Inherited Retinal Dystrophy in Southeastern United States: Characterization of South Carolina Patients and Comparative Literature Review. Genes. 2022; 13(8):1490. https://doi.org/10.3390/genes13081490
Chicago/Turabian StyleGriffith, Joseph, III, Kareem Sioufi, Laurie Wilbanks, George N. Magrath, Emil A. T. Say, Michael J. Lyons, Meg Wilkes, Gurpur Shashidhar Pai, and Mae Millicent Winfrey Peterseim. 2022. "Inherited Retinal Dystrophy in Southeastern United States: Characterization of South Carolina Patients and Comparative Literature Review" Genes 13, no. 8: 1490. https://doi.org/10.3390/genes13081490
APA StyleGriffith, J., III, Sioufi, K., Wilbanks, L., Magrath, G. N., Say, E. A. T., Lyons, M. J., Wilkes, M., Pai, G. S., & Peterseim, M. M. W. (2022). Inherited Retinal Dystrophy in Southeastern United States: Characterization of South Carolina Patients and Comparative Literature Review. Genes, 13(8), 1490. https://doi.org/10.3390/genes13081490