
Comparative Transcriptomics and Methylomics Reveal Adaptive Responses of Digestive and Metabolic Genes to Dietary Shift in Giant and Red Pandas

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Library Preparation and RNA Sequencing
2.3. RNA-Seq Read Mapping
2.4. RNA-Seq Analysis
2.5. Library Preparation and Bisulfite Sequencing
2.6. BS-Seq Read Mapping and Data Processing
2.7. Promoter Methylation Analysis
3. Results
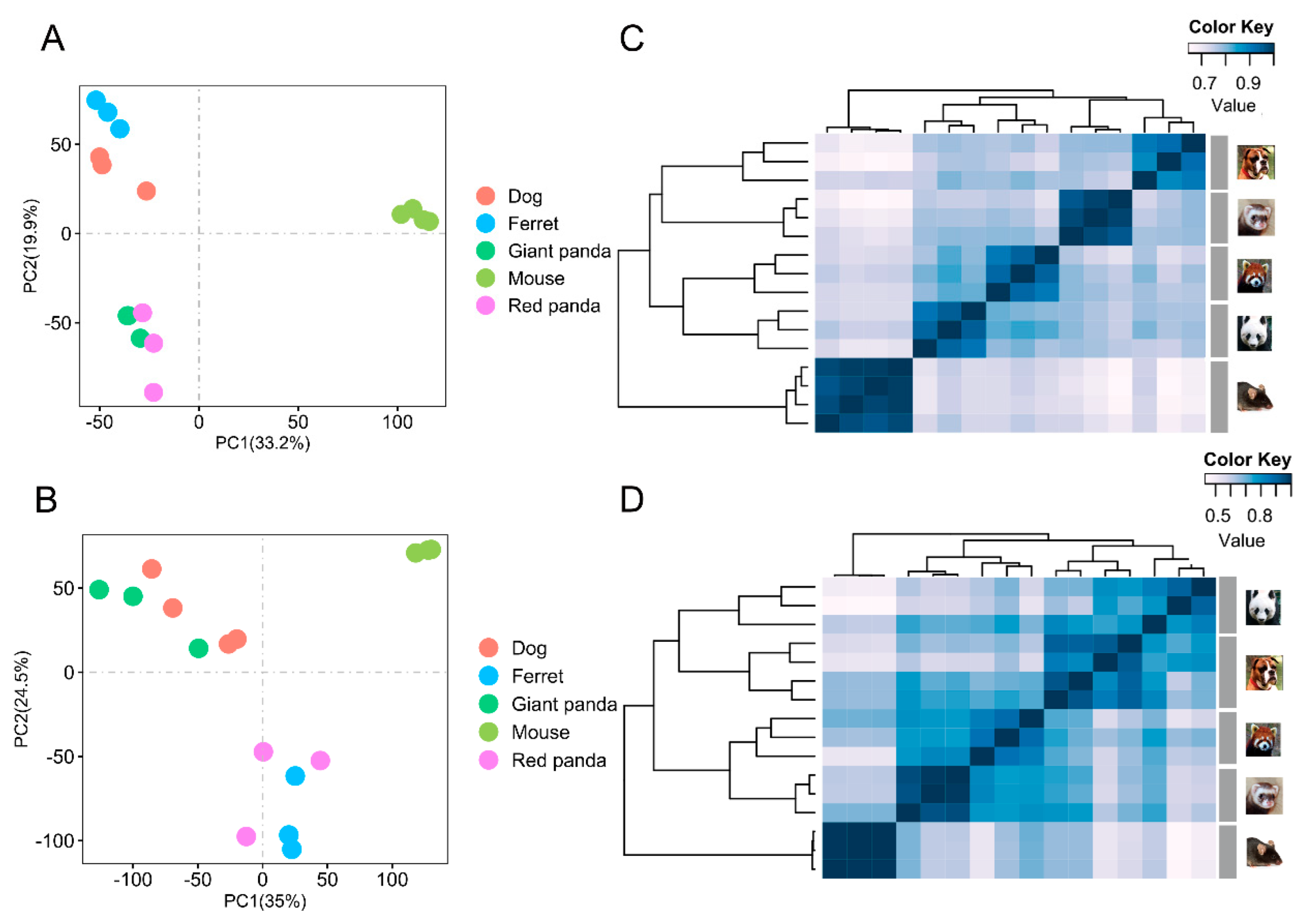
3.1. Transcriptomic PCA and Clustering Analyses
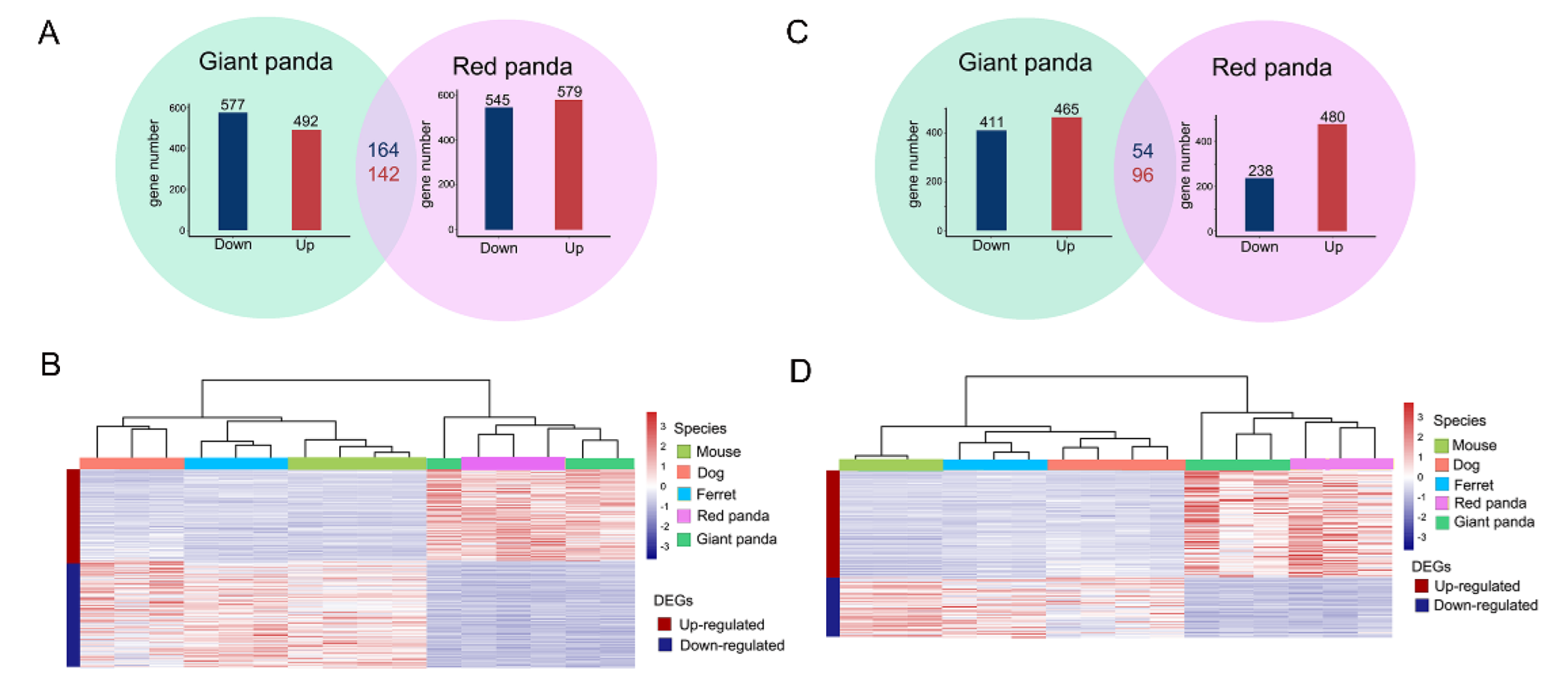
3.2. Transcriptomic Profiles of the Stomach
3.3. Transcriptomic Profiles of the Small Intestine
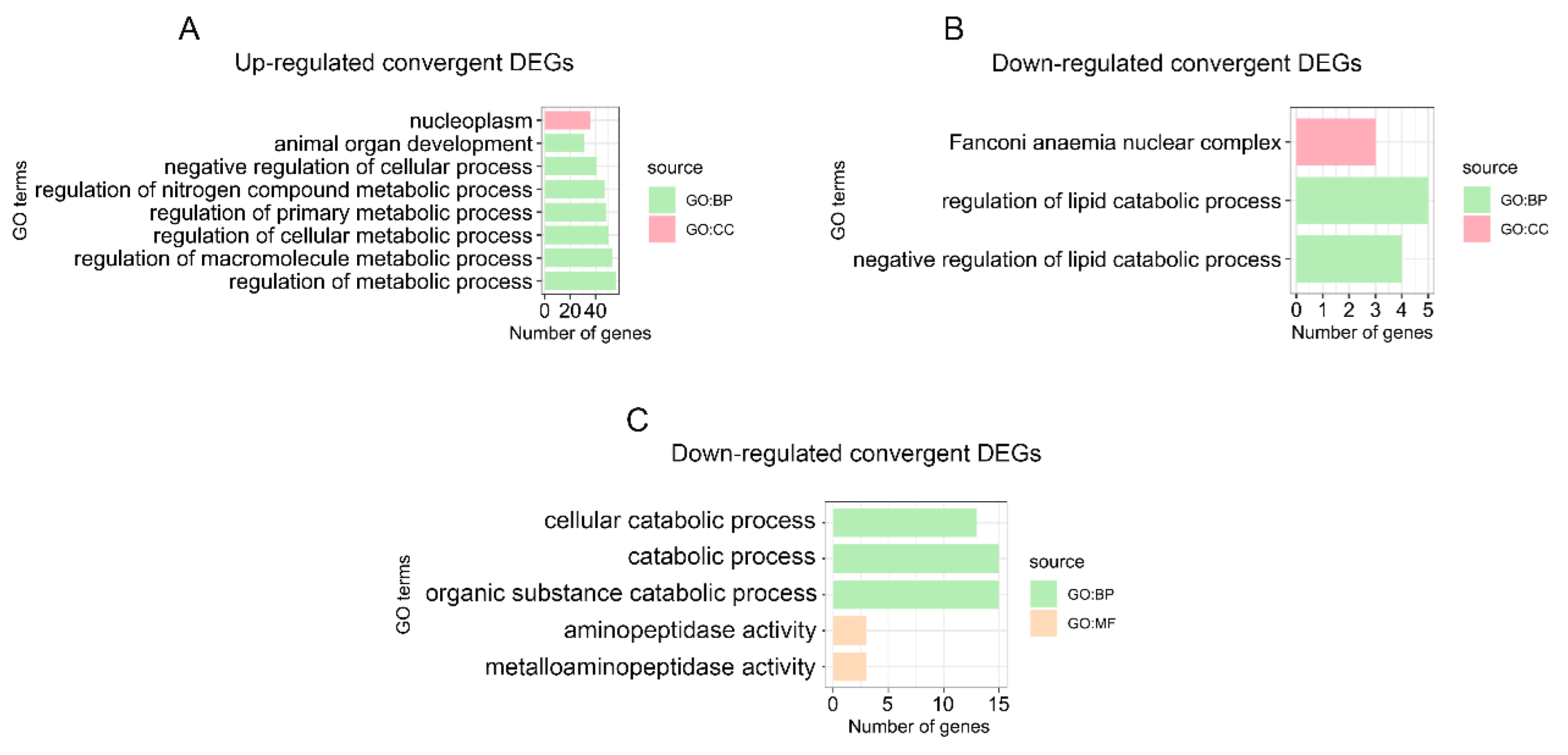
3.4. Enrichment Analysis of Convergent DEGs and Identification of Digestion- and Metabolism-Related Convergent DEGs in the Stomach
3.5. Enrichment Analysis of Convergent DEGs and Identification of Digestion- and Metabolism-Related Convergent DEGs in the Small Intestine
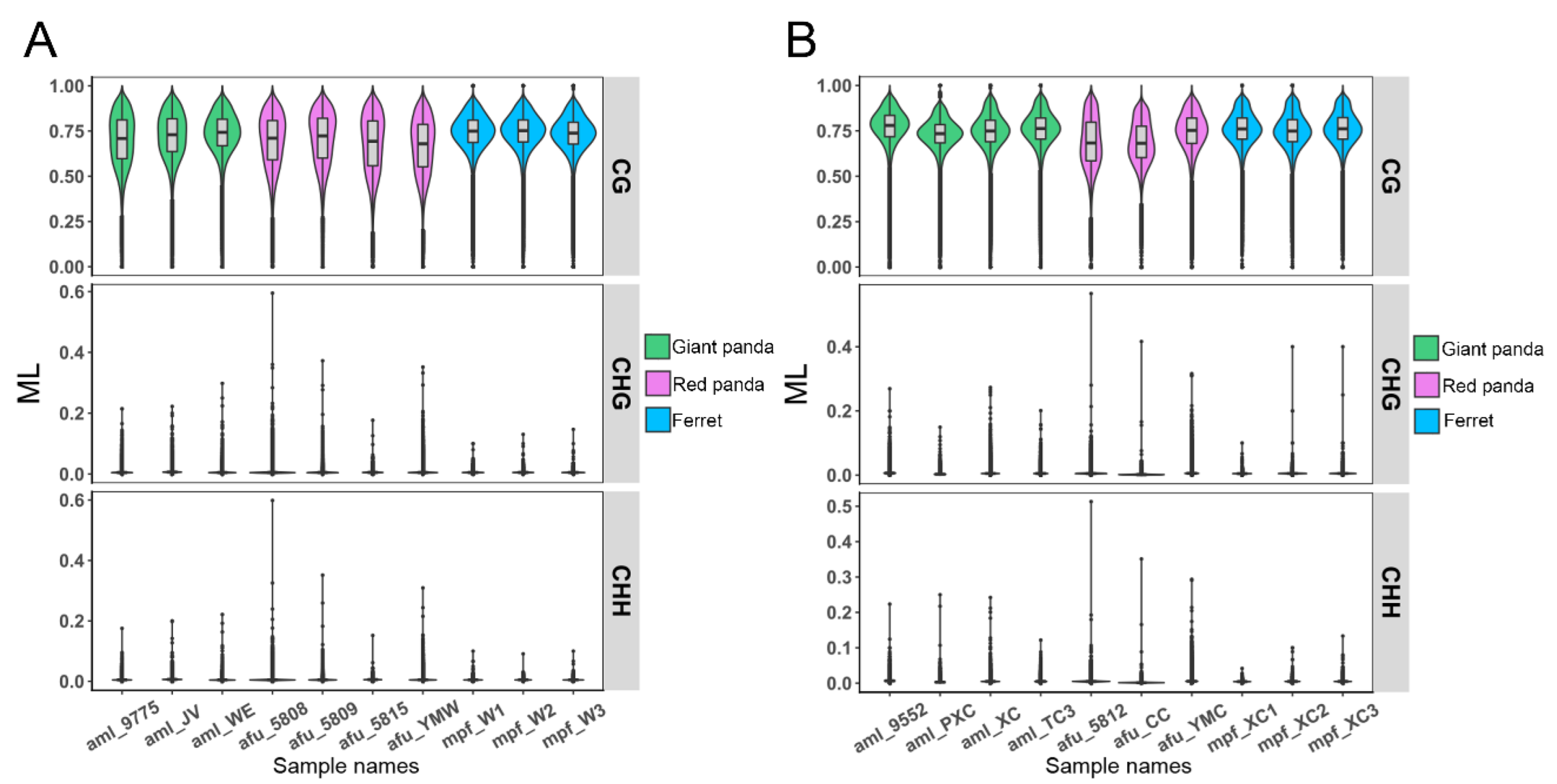
3.6. DNA Methylation Patterns in Genomes
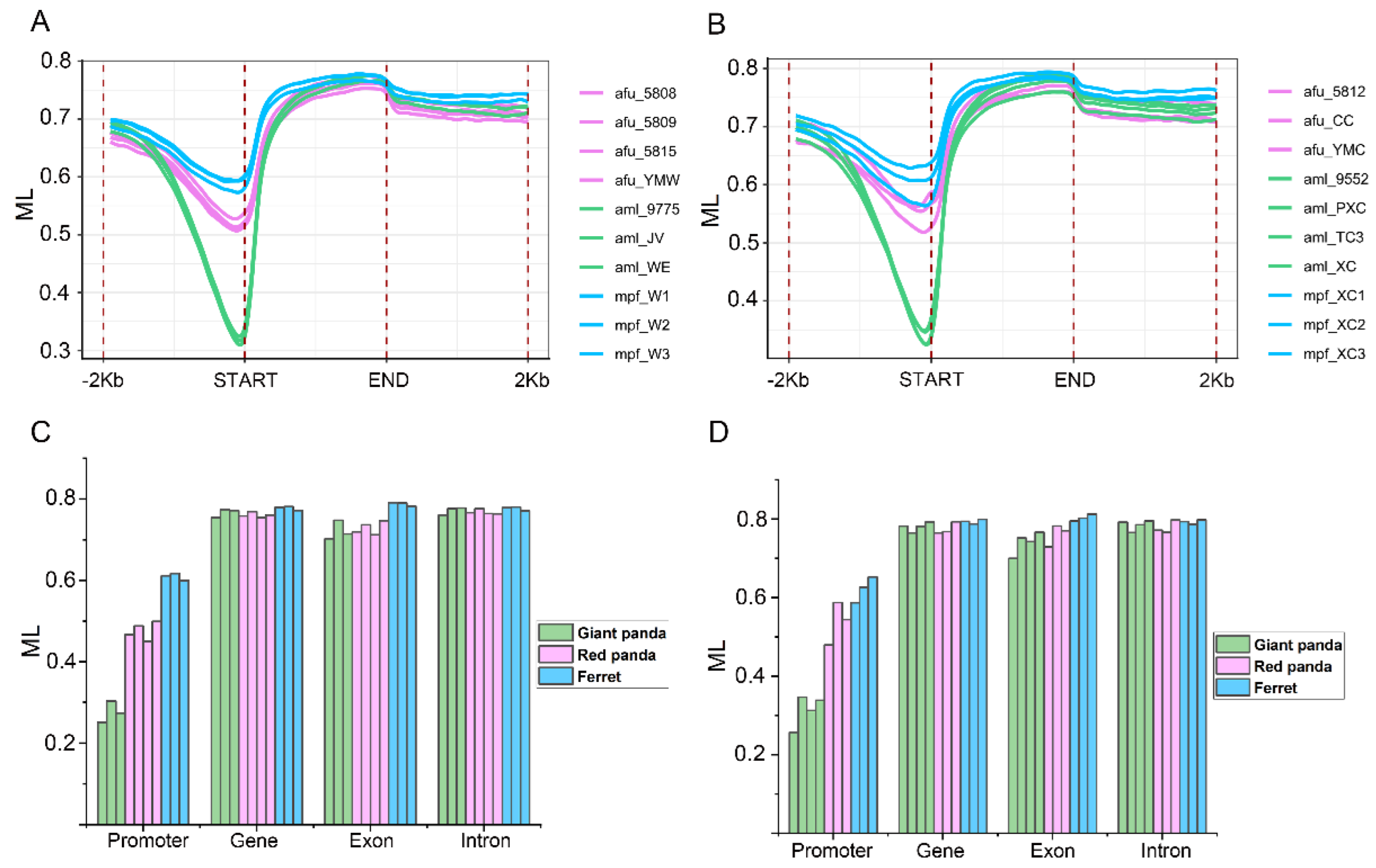
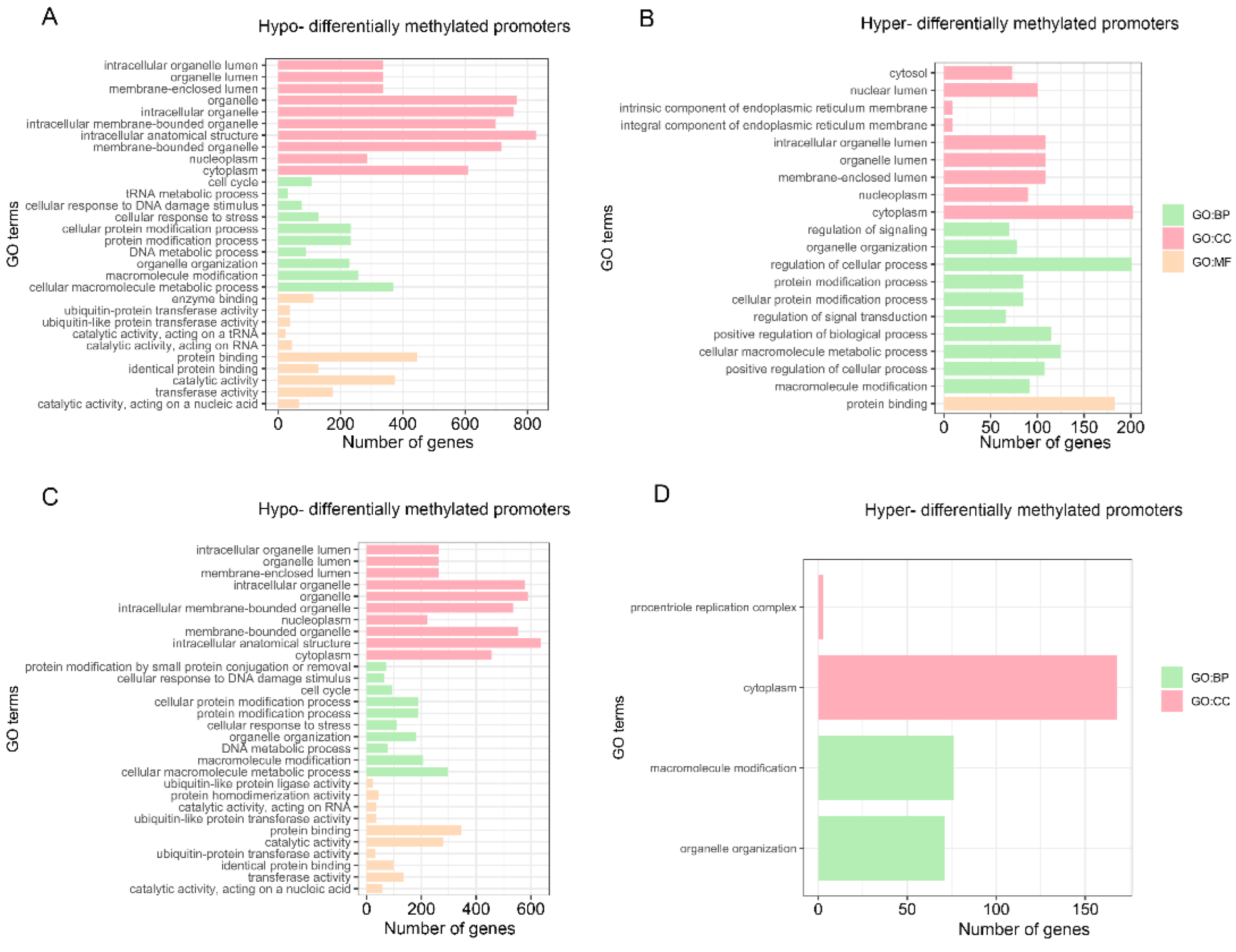
3.7. Promoter Methylation Profiles
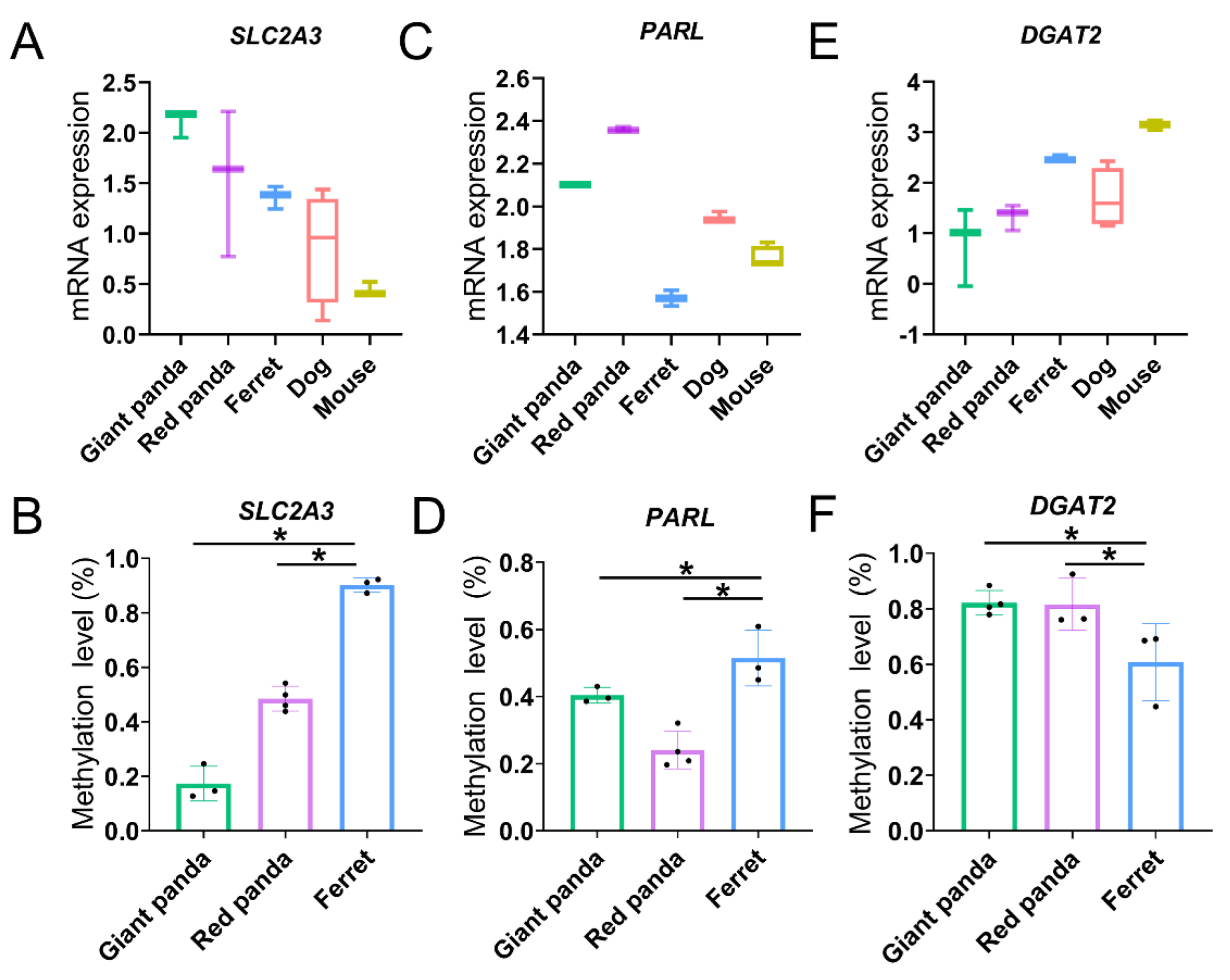
3.8. Methylation Levels of Convergent Promoters
4. Discussion
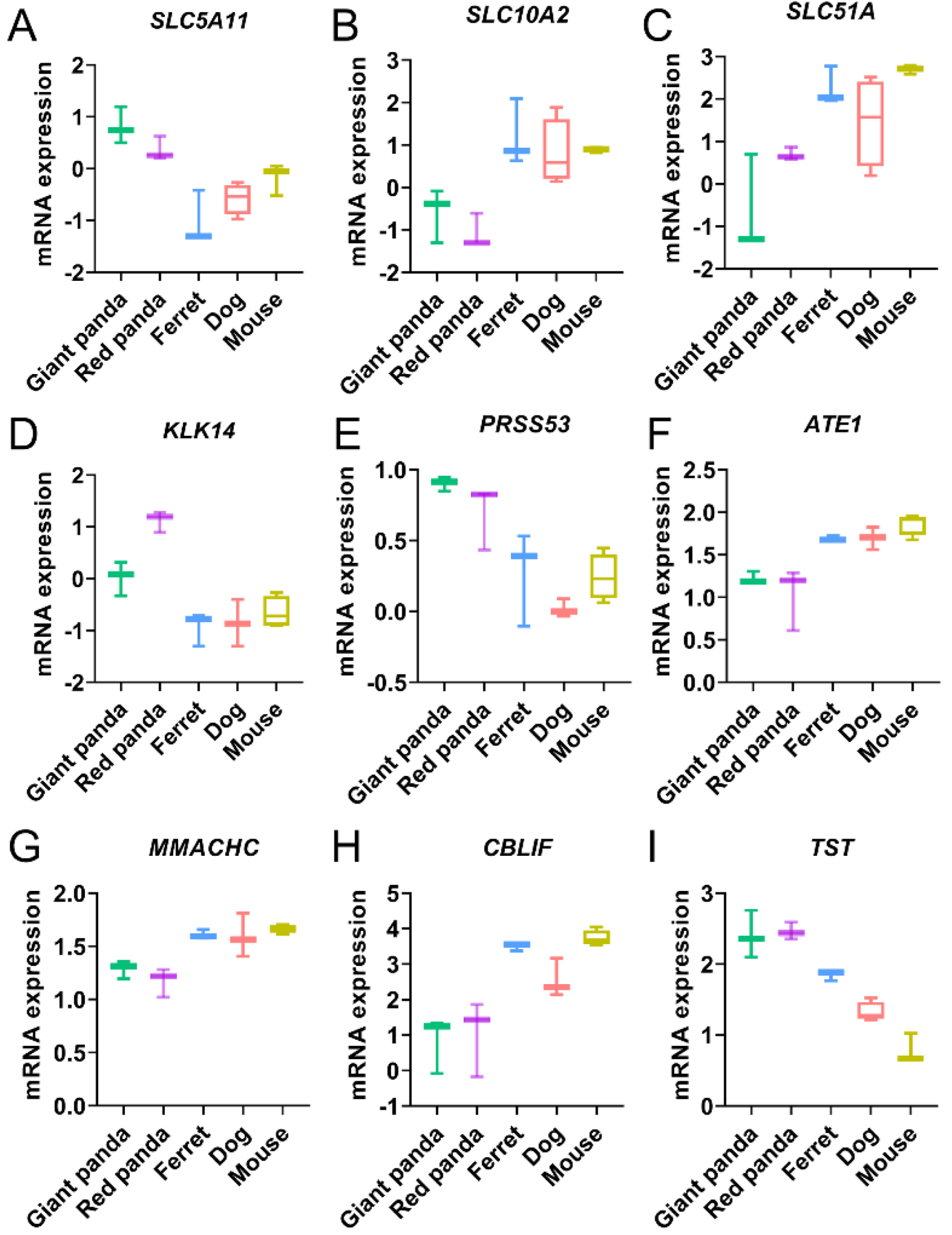
4.1. Convergent DEG Related to Carbohydrate Utilization
4.2. Convergent DEGs Related to Bile Secretion
4.3. Convergent DEGs Related to Amino Acid Metabolism
4.4. Convergent DEGs Related to Vitamin Utilization and Cyanide Detoxification
4.5. DNA Methylation Regulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, K.; Liu, Q.; Xu, D.M.; Qi, F.Y.; Bai, J.; He, S.W.; Chen, P.; Zhou, X.; Cai, W.Z.; Chen, Z.Z.; et al. Echolocation in soft-furred tree mice. Science 2021, 372, 6548. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, H. Convergent evolution of bird-mammal shared characteristics for adapting to nocturnality. Proc. Biol. Sci. 2019, 286, 20182185. [Google Scholar] [CrossRef]
- Christin, P.A.; Weinreich, D.M.; Besnard, G. Causes and evolutionary significance of genetic convergence. Trends Genet. TIG 2010, 26, 400–405. [Google Scholar] [CrossRef]
- Stern, D.L. The genetic causes of convergent evolution. Nat. Rev. Genet. 2013, 14, 751–764. [Google Scholar] [CrossRef]
- Storz, J.F. Causes of molecular convergence and parallelism in protein evolution. Nat. Rev. Genet. 2016, 17, 239–250. [Google Scholar] [CrossRef] [PubMed]
- de Sousa-Pereira, P.; Cova, M.; Abrantes, J.; Ferreira, R.; Trindade, F.; Barros, A.; Gomes, P.; Colaco, B.; Amado, F.; Esteves, P.J.; et al. Cross-species comparison of mammalian saliva using an LC-MALDI based proteomic approach. Proteomics 2015, 15, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.A.; Rogers, Q.R.; Morris, J.G. Methionine requirement of kittens given amino acid diets containing adequate cystine. Br. J. Nutr. 1983, 49, 411–417. [Google Scholar] [CrossRef]
- Wang, B.J.; Xia, J.M.; Wang, Q.; Yu, J.L.; Song, Z.; Zhao, H. Diet and Adaptive Evolution of Alanine-Glyoxylate Aminotransferase Mitochondrial Targeting in Birds. Mol. Biol. Evol. 2020, 37, 786–798. [Google Scholar] [CrossRef]
- Hu, Y.; Wu, Q.; Ma, S.; Ma, T.; Shan, L.; Wang, X.; Nie, Y.; Ning, Z.; Yan, L.; Xiu, Y.; et al. Comparative genomics reveals convergent evolution between the bamboo-eating giant and red pandas. Proc. Natl. Acad. Sci. USA 2017, 114, 1081–1086. [Google Scholar] [CrossRef]
- Anton, M.; Salesa, M.J.; Pastor, J.F.; Peigne, S.; Morales, J. Implications of the functional anatomy of the hand and forearm of Ailurus fulgens (Carnivora, Ailuridae) for the evolution of the ‘false-thumb’ in pandas. J. Anat. 2006, 209, 757–764. [Google Scholar] [CrossRef]
- Figueirido, B.; Serrano-Alarcon, F.J.; Slater, G.J.; Palmqvist, P. Shape at the cross-roads: Homoplasy and history in the evolution of the carnivoran skull towards herbivory. J. Evol. Biol. 2010, 23, 2579–2594. [Google Scholar] [CrossRef] [PubMed]
- Figueirido, B.; Tseng, Z.J.; Martin-Serra, A. Skull shape evolution in durophagous carnivorans. Evol. Int. J. Org. Evol. 2013, 67, 1975–1993. [Google Scholar] [CrossRef] [PubMed]
- Figueirido, B.; Tseng, Z.J.; Serrano-Alarcon, F.J.; Martin-Serra, A.; Pastor, J.F. Three-dimensional computer simulations of feeding behaviour in red and giant pandas relate skull biomechanics with dietary niche partitioning. Biol. Lett. 2014, 10, 20140196. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Wang, X.; Hu, Y.; Wu, Q.; Nie, Y.; Dong, J.; Ding, Y.; Yan, L.; Wei, F. Diet drives convergent evolution of gut microbiomes in bamboo-eating species. Sci. China Life Sci. 2021, 64, 88–95. [Google Scholar] [CrossRef]
- Knott, K.K.; Christian, A.L.; Falcone, J.F.; Vance, C.K.; Bauer, L.L.; Fahey, G.C., Jr.; Kouba, A.J. Phenological changes in bamboo carbohydrates explain the preference for culm over leaves by giant pandas (Ailuropoda melanoleuca) during spring. PLoS ONE 2017, 12, e0177582. [Google Scholar] [CrossRef]
- Shan, L.; Wu, Q.; Wang, L.; Zhang, L.; Wei, F. Lineage-specific evolution of bitter taste receptor genes in the giant and red pandas implies dietary adaptation. Integr. Zool. 2018, 13, 152–159. [Google Scholar] [CrossRef]
- Yao, R.; Yang, Z.; Zhang, Z.; Hu, T.; Chen, H.; Huang, F.; Gu, X.; Yang, X.; Lu, G.; Zhu, L. Are the gut microbial systems of giant pandas unstable? Heliyon 2019, 5, e02480. [Google Scholar] [CrossRef]
- Choudhury, D.; Sahu, J.K.; Sharma, G.D. Value addition to bamboo shoots: A review. J. Food Sci. Technol. 2012, 49, 407–414. [Google Scholar] [CrossRef]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef]
- Smith, Z.D.; Chan, M.M.; Mikkelsen, T.S.; Gu, H.; Gnirke, A.; Regev, A.; Meissner, A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 2012, 484, 339–344. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Hernando-Herraez, I.; Garcia-Perez, R.; Sharp, A.J.; Marques-Bonet, T. DNA Methylation: Insights into Human Evolution. PLoS Genet. 2015, 11, e1005661. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef]
- Smid, M.; Coebergh van den Braak, R.R.J.; van de Werken, H.J.G.; van Riet, J.; van Galen, A.; de Weerd, V.; van der Vlugt-Daane, M.; Bril, S.I.; Lalmahomed, Z.S.; Kloosterman, W.P.; et al. Gene length corrected trimmed mean of M-values (GeTMM) processing of RNA-seq data performs similarly in intersample analyses while improving intrasample comparisons. BMC Bioinform. 2018, 19, 236. [Google Scholar] [CrossRef]
- Kerr, M.K.; Martin, M.; Churchill, G.A. Analysis of variance for gene expression microarray data. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2000, 7, 819–837. [Google Scholar] [CrossRef]
- Bourgon, R.; Gentleman, R.; Huber, W. Independent filtering increases detection power for high-throughput experiments. Proc. Natl. Acad. Sci. USA 2010, 107, 9546–9551. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Barton, L.; Bures, D.; Rehak, D.; Kott, T.; Makovicky, P. Tissue-specific fatty acid composition, cellularity, and gene expression in diverse cattle breeds. Anim. Int. J. Anim. Biosci. 2021, 15, 100025. [Google Scholar] [CrossRef]
- Kryuchkova-Mostacci, N.; Robinson-Rechavi, M. A benchmark of gene expression tissue-specificity metrics. Brief. Bioinform. 2017, 18, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, F.; Feng, X.; Yang, H.; Zhu, A.; Pang, J.; Han, L.; Zhang, T.; Yao, X.; Wang, F. Genome-wide analysis of DNA Methylation profiles on sheep ovaries associated with prolificacy using whole-genome Bisulfite sequencing. BMC Genom. 2017, 18, 759. [Google Scholar] [CrossRef]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, L.; Yang, Q.; Yan, C.; Jiang, P.; Lan, Y.; Wang, J.; Tang, R.; He, M.; Lei, G.; et al. Age-related gene expression and DNA methylation changes in rhesus macaque. Genomics 2020, 112, 5147–5156. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Y.; Zhou, G.; Gao, Y.; Ma, S.; Chen, Y.; Song, J.; Wang, X. Whole-genome bisulfite sequencing of goat skins identifies signatures associated with hair cycling. BMC Genom. 2018, 19, 638. [Google Scholar] [CrossRef]
- Sevane, N.; Martinez, R.; Bruford, M.W. Genome-wide differential DNA methylation in tropically adapted Creole cattle and their Iberian ancestors. Anim. Genet. 2019, 50, 15–26. [Google Scholar] [CrossRef]
- Arechederra, M.; Daian, F.; Yim, A.; Bazai, S.K.; Richelme, S.; Dono, R.; Saurin, A.J.; Habermann, B.H.; Maina, F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018, 9, 3164. [Google Scholar] [CrossRef] [PubMed]
- Dierenfeld, E.S.; Hintz, H.F.; Robertson, J.B.; Van Soest, P.J.; Oftedal, O.T. Utilization of bamboo by the giant panda. J. Nutr. 1982, 112, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.; Hou, R.; Spotila, J.R.; Paladino, F.V.; Qi, D.; Zhang, Z. Metabolic rate of the red panda, Ailurus fulgens, a dietary bamboo specialist. PLoS ONE 2017, 12, e0173274. [Google Scholar] [CrossRef]
- Ugrankar, R.; Theodoropoulos, P.; Akdemir, F.; Henne, W.M.; Graff, J.M. Circulating glucose levels inversely correlate with Drosophila larval feeding through insulin signaling and SLC5A11. Commun. Biol. 2018, 1, 110. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, M.; Scow, R.O. Lingual lipase and its role in the digestion of dietary lipid. J. Clin. Investig. 1973, 52, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Valderrama, J.; Wilde, P.; Macierzanka, A.; Mackie, A. The role of bile salts in digestion. Adv. Colloid Interface Sci. 2011, 165, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Dong, Y.Q.; Jia, G.X.; Fan, S.M.; Li, S.Z.; Yang, S.S.; Li, Y.B. ASBT(SLC10A2): A promising target for treatment of diseases and drug discovery. Biomed. Pharmacother. 2020, 132, 110835. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N.; Christian, W.V.; Wheeler, S.G.; Hammond, C.L. The heteromeric organic solute transporter, OSTalpha-OSTbeta/SLC51: A transporter for steroid-derived molecules. Mol. Asp. Med. 2013, 34, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Xu, E.; Chen, C.; Fu, J.; Zhu, L.; Shu, J.; Jin, M.; Wang, Y.; Zong, X. Dietary fatty acids in gut health: Absorption, metabolism and function. Anim. Nutr. 2021, 7, 1337–1344. [Google Scholar] [CrossRef]
- Hofmann, A.F.; Hagey, L.R. Key discoveries in bile acid chemistry and biology and their clinical applications: History of the last eight decades. J. Lipid Res. 2014, 55, 1553–1595. [Google Scholar] [CrossRef]
- D’Aquila, T.; Hung, Y.H.; Carreiro, A.; Buhman, K.K. Recent discoveries on absorption of dietary fat: Presence, synthesis, and metabolism of cytoplasmic lipid droplets within enterocytes. Biochim. Biophys. Acta 2016, 1861, 730–747. [Google Scholar] [CrossRef] [PubMed]
- Rushton, D.H. Nutritional factors and hair loss. Clin. Exp. Dermatol. 2002, 27, 396–404. [Google Scholar] [CrossRef]
- Ghosh, S.; Smriga, M.; Vuvor, F.; Suri, D.; Mohammed, H.; Armah, S.M.; Scrimshaw, N.S. Effect of lysine supplementation on health and morbidity in subjects belonging to poor peri-urban households in Accra, Ghana. Am. J. Clin. Nutr. 2010, 92, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Melik, Z.; Zaletel, P.; Virtic, T.; Cankar, K. L-arginine as dietary supplement for improving microvascular function. Clin. Hemorheol. Microcirc. 2017, 65, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Cylwik, D.; Mogielnicki, A.; Buczko, W. L-arginine and cardiovascular system. Pharmacol. Rep. 2005, 57, 14–22. [Google Scholar] [PubMed]
- Wang, J.; Han, X.; Wong, C.C.; Cheng, H.; Aslanian, A.; Xu, T.; Leavis, P.; Roder, H.; Hedstrom, L.; Yates, J.R., 3rd; et al. Arginyltransferase ATE1 catalyzes midchain arginylation of proteins at side chain carboxylates in vivo. Chem. Biol. 2014, 21, 331–337. [Google Scholar] [CrossRef]
- Herrmann, W.; Geisel, J. Vegetarian lifestyle and monitoring of vitamin B-12 status. Clin. Chim. Acta Int. J. Clin. Chem. 2002, 326, 47–59. [Google Scholar] [CrossRef]
- Fang, H.; Kang, J.; Zhang, D. Microbial production of vitamin B12: A review and future perspectives. Microb. Cell Factories 2017, 16, 15. [Google Scholar] [CrossRef]
- O’Leary, F.; Samman, S. Vitamin B12 in health and disease. Nutrients 2010, 2, 299–316. [Google Scholar] [CrossRef] [PubMed]
- Froese, D.S.; Kopec, J.; Fitzpatrick, F.; Schuller, M.; McCorvie, T.J.; Chalk, R.; Plessl, T.; Fettelschoss, V.; Fowler, B.; Baumgartner, M.R.; et al. Structural Insights into the MMACHC-MMADHC Protein Complex Involved in Vitamin B12 Trafficking. J. Biol. Chem. 2015, 290, 29167–29177. [Google Scholar] [CrossRef]
- Huang, H.; Yie, S.; Liu, Y.; Wang, C.; Cai, Z.; Zhang, W.; Lan, J.; Huang, X.; Luo, L.; Cai, K.; et al. Dietary resources shape the adaptive changes of cyanide detoxification function in giant panda (Ailuropoda melanoleuca). Sci. Rep. 2016, 6, 34700. [Google Scholar] [CrossRef] [PubMed]
- Sani, M.; Gadacha, W.; Boughattas, N.A.; Reinberg, A.; Ben Attia, M. Circadian and ultradian (12 h) rhythms of hepatic thiosulfate sulfurtransferase (rhodanese) activity in mice during the first two months of life. Chronobiol. Int. 2006, 23, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Billaut-Laden, I.; Allorge, D.; Crunelle-Thibaut, A.; Rat, E.; Cauffiez, C.; Chevalier, D.; Houdret, N.; Lo-Guidice, J.M.; Broly, F. Evidence for a functional genetic polymorphism of the human thiosulfate sulfurtransferase (Rhodanese), a cyanide and H2S detoxification enzyme. Toxicology 2006, 225, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ashe, A.; Colot, V.; Oldroyd, B.P. How does epigenetics influence the course of evolution? Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2021, 376, 20200111. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.V.; Goodisman, M.A. Computational approaches for understanding the evolution of DNA methylation in animals. Epigenetics 2009, 4, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Wachter, E.; Quante, T.; Merusi, C.; Arczewska, A.; Stewart, F.; Webb, S.; Bird, A. Synthetic CpG islands reveal DNA sequence determinants of chromatin structure. eLife 2014, 3, e03397. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.R.; Dessus-Babus, S.; Burger, L.; Schubeler, D. High-throughput engineering of a mammalian genome reveals building principles of methylation states at CG rich regions. eLife 2014, 3, e04094. [Google Scholar] [CrossRef] [PubMed]
- Moarii, M.; Boeva, V.; Vert, J.P.; Reyal, F. Changes in correlation between promoter methylation and gene expression in cancer. BMC Genom. 2015, 16, 873. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Hernando-Herraez, I.; Prado-Martinez, J.; Garg, P.; Fernandez-Callejo, M.; Heyn, H.; Hvilsom, C.; Navarro, A.; Esteller, M.; Sharp, A.J.; Marques-Bonet, T. Dynamics of DNA methylation in recent human and great ape evolution. PLoS Genet. 2013, 9, e1003763. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H.; Zhou, D.; Moody, L.; Lezmi, S.; Chen, H.; Pan, Y.X. High-fat diet caused widespread epigenomic differences on hepatic methylome in rat. Physiol. Genom. 2015, 47, 514–523. [Google Scholar] [CrossRef]
- Seatter, M.J.; De la Rue, S.A.; Porter, L.M.; Gould, G.W. QLS motif in transmembrane helix VII of the glucose transporter family interacts with the C-1 position of D-glucose and is involved in substrate selection at the exofacial binding site. Biochemistry 1998, 37, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.C.; Steinhauser, C.B.; Gao, H.; Seo, H.; McLendon, B.A.; Burghardt, R.C.; Wu, G.; Bazer, F.W.; Johnson, G.A. Steroids Regulate SLC2A1 and SLC2A3 to Deliver Glucose Into Trophectoderm for Metabolism via Glycolysis. Endocrinology 2020, 161. [Google Scholar] [CrossRef]
- van Tilbeurgh, H.; Bezzine, S.; Cambillau, C.; Verger, R.; Carriere, F. Colipase: Structure and interaction with pancreatic lipase. Biochim. Biophys. Acta 1999, 1441, 173–184. [Google Scholar] [CrossRef]
- Gallo, L.L.; Clark, S.B.; Myers, S.; Vahouny, G.V. Cholesterol absorption in rat intestine: Role of cholesterol esterase and acyl coenzyme A:cholesterol acyltransferase. J. Lipid Res. 1984, 25, 604–612. [Google Scholar] [CrossRef]
- Yen, C.E.; Nelson, D.W.; Yen, M.I. Intestinal triacylglycerol synthesis in fat absorption and systemic energy metabolism. J. Lipid Res. 2015, 56, 489–501. [Google Scholar] [CrossRef]
- Hung, Y.H.; Carreiro, A.L.; Buhman, K.K. Dgat1 and Dgat2 regulate enterocyte triacylglycerol distribution and alter proteins associated with cytoplasmic lipid droplets in response to dietary fat. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 600–614. [Google Scholar] [CrossRef]
- Carone, B.R.; Fauquier, L.; Habib, N.; Shea, J.M.; Hart, C.E.; Li, R.; Bock, C.; Li, C.; Gu, H.; Zamore, P.D.; et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 2010, 143, 1084–1096. [Google Scholar] [CrossRef]
- Trumpff, C.; Sturm, G.; Picard, M.; Foss, S.; Lee, S.; Feng, T.; Cardenas, A.; McCormack, C.; Champagne, F.A.; Monk, C. Added sugar intake during pregnancy: Fetal behavior, birth outcomes, and placental DNA methylation. Dev. Psychobiol. 2021, 63, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.L.; Dong, S.; Gao, L.F.; Li, L.; Xi, Y.D.; Ma, W.W.; Yuan, L.H.; Xiao, R. Global DNA methylation was changed by a maternal high-lipid, high-energy diet during gestation and lactation in male adult mice liver. Br. J. Nutr. 2015, 113, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Altmann, S.; Murani, E.; Schwerin, M.; Metges, C.C.; Wimmers, K.; Ponsuksili, S. Dietary protein restriction and excess of pregnant German Landrace sows induce changes in hepatic gene expression and promoter methylation of key metabolic genes in the offspring. J. Nutr. Biochem. 2013, 24, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, K.D.; Allegrucci, C.; Singh, R.; Gardner, D.S.; Sebastian, S.; Bispham, J.; Thurston, A.; Huntley, J.F.; Rees, W.D.; Maloney, C.A.; et al. DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proc. Natl. Acad. Sci. USA 2007, 104, 19351–19356. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Shen, F.; Jie, X.; Zhang, L.; Yan, G.; Wu, H.; Huang, Y.; Hou, R.; Yue, B.; Zhang, X. Comparative Transcriptomics and Methylomics Reveal Adaptive Responses of Digestive and Metabolic Genes to Dietary Shift in Giant and Red Pandas. Genes 2022, 13, 1446. https://doi.org/10.3390/genes13081446
Li L, Shen F, Jie X, Zhang L, Yan G, Wu H, Huang Y, Hou R, Yue B, Zhang X. Comparative Transcriptomics and Methylomics Reveal Adaptive Responses of Digestive and Metabolic Genes to Dietary Shift in Giant and Red Pandas. Genes. 2022; 13(8):1446. https://doi.org/10.3390/genes13081446
Chicago/Turabian StyleLi, Lu, Fujun Shen, Xiaodie Jie, Liang Zhang, Guoqiang Yan, Honglin Wu, Yan Huang, Rong Hou, Bisong Yue, and Xiuyue Zhang. 2022. "Comparative Transcriptomics and Methylomics Reveal Adaptive Responses of Digestive and Metabolic Genes to Dietary Shift in Giant and Red Pandas" Genes 13, no. 8: 1446. https://doi.org/10.3390/genes13081446
APA StyleLi, L., Shen, F., Jie, X., Zhang, L., Yan, G., Wu, H., Huang, Y., Hou, R., Yue, B., & Zhang, X. (2022). Comparative Transcriptomics and Methylomics Reveal Adaptive Responses of Digestive and Metabolic Genes to Dietary Shift in Giant and Red Pandas. Genes, 13(8), 1446. https://doi.org/10.3390/genes13081446