
CLN8 Gene Compound Heterozygous Variants: A New Case and Protein Bioinformatics Analyses

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
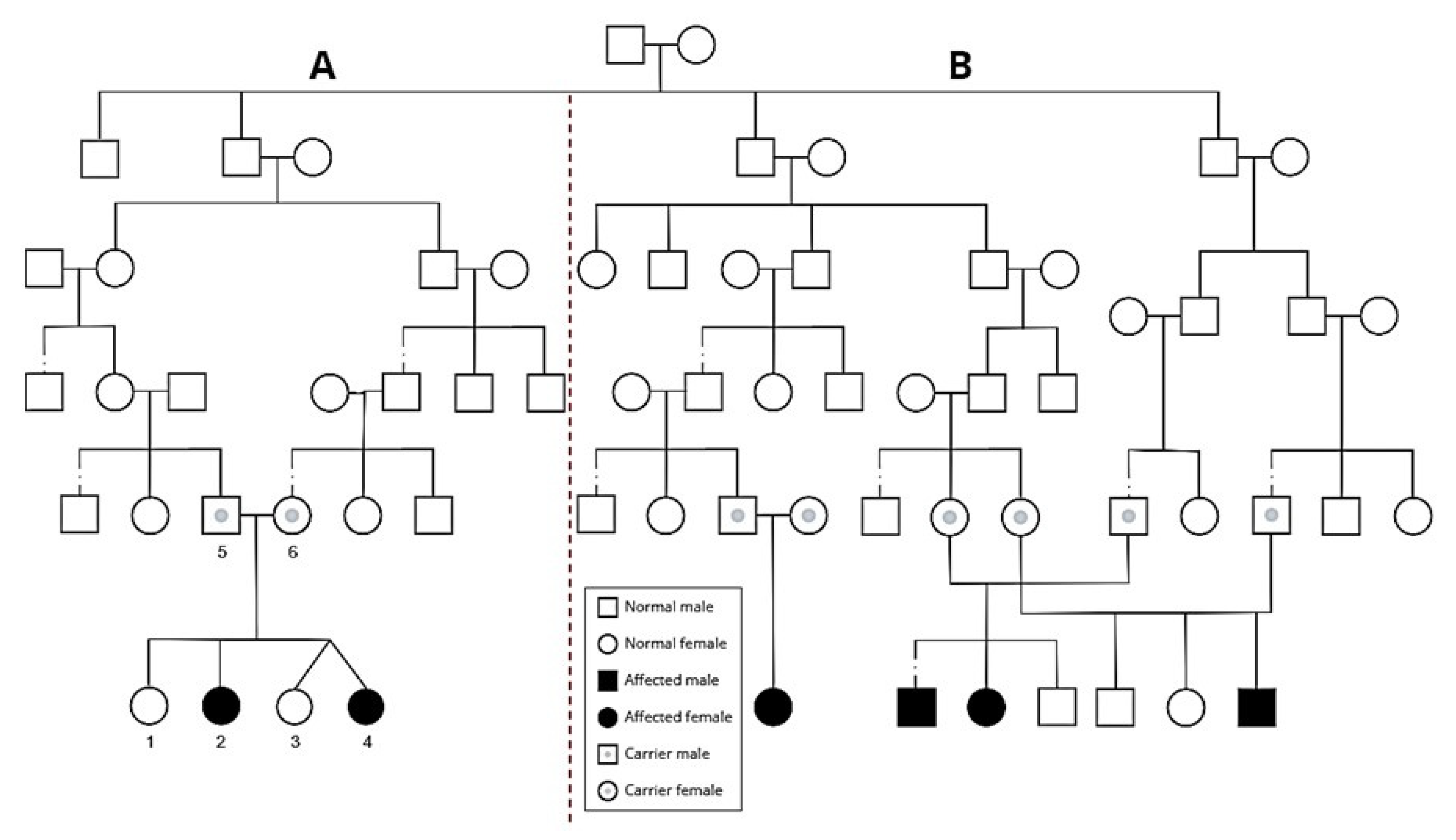
2.1. Clinical Description
2.1.1. Patient A
2.1.2. Patient B
2.2. Genetic Analysis
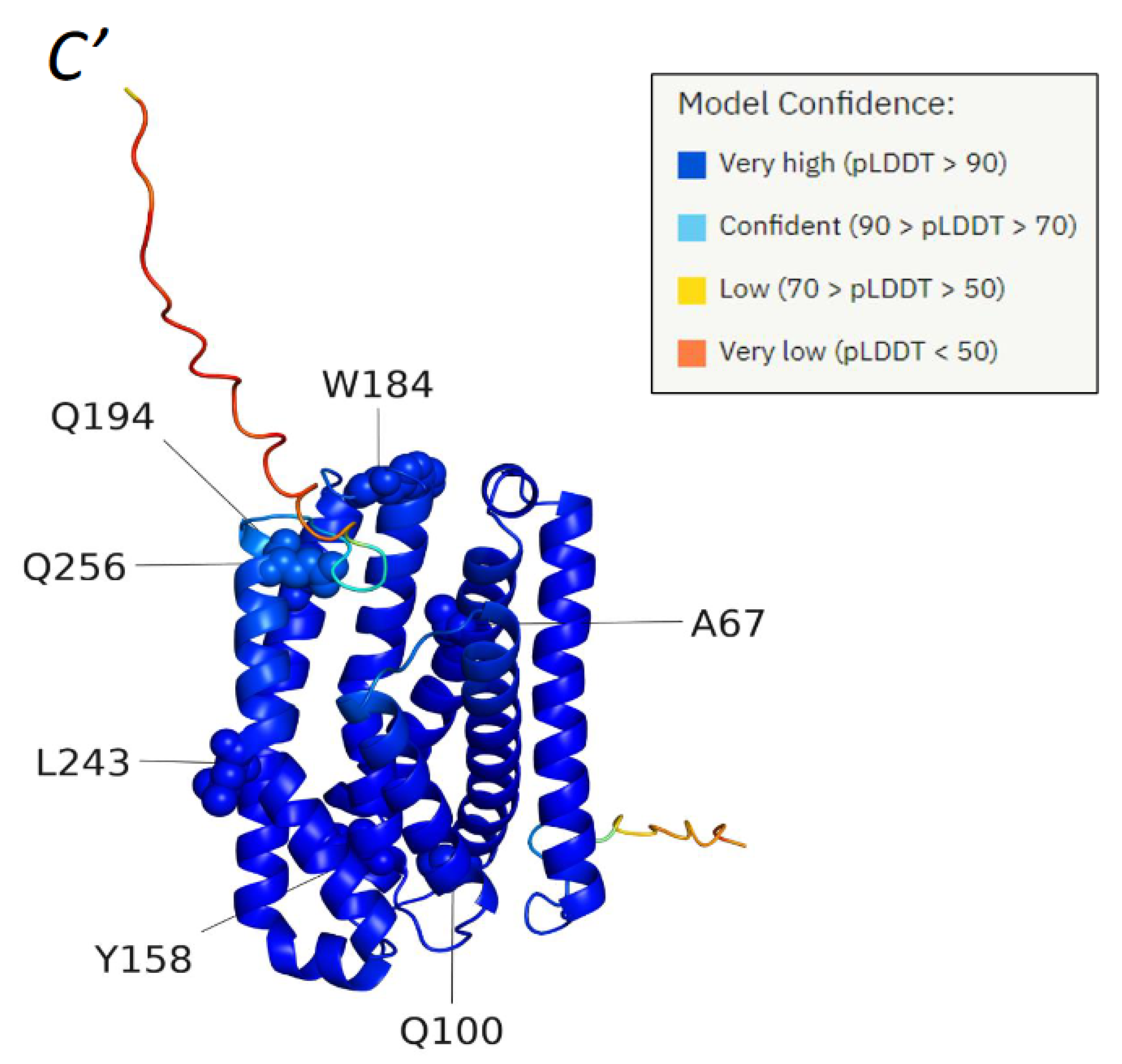
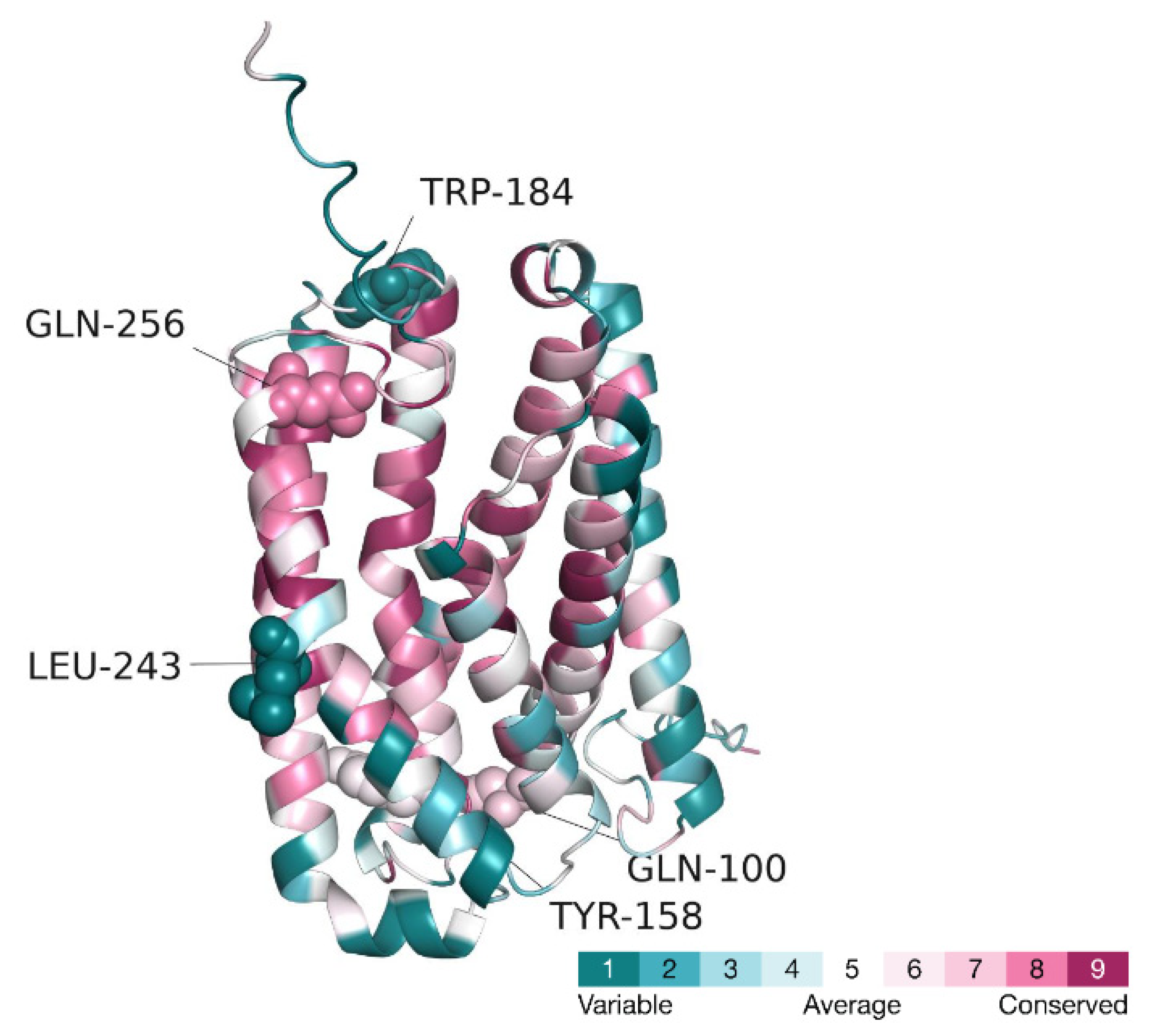
2.3. Structural Bioinformatic Analysis
3. Results
3.1. Mutational Analysis of the CLN8 Gene Product
3.1.1. Variants Found by Our Study
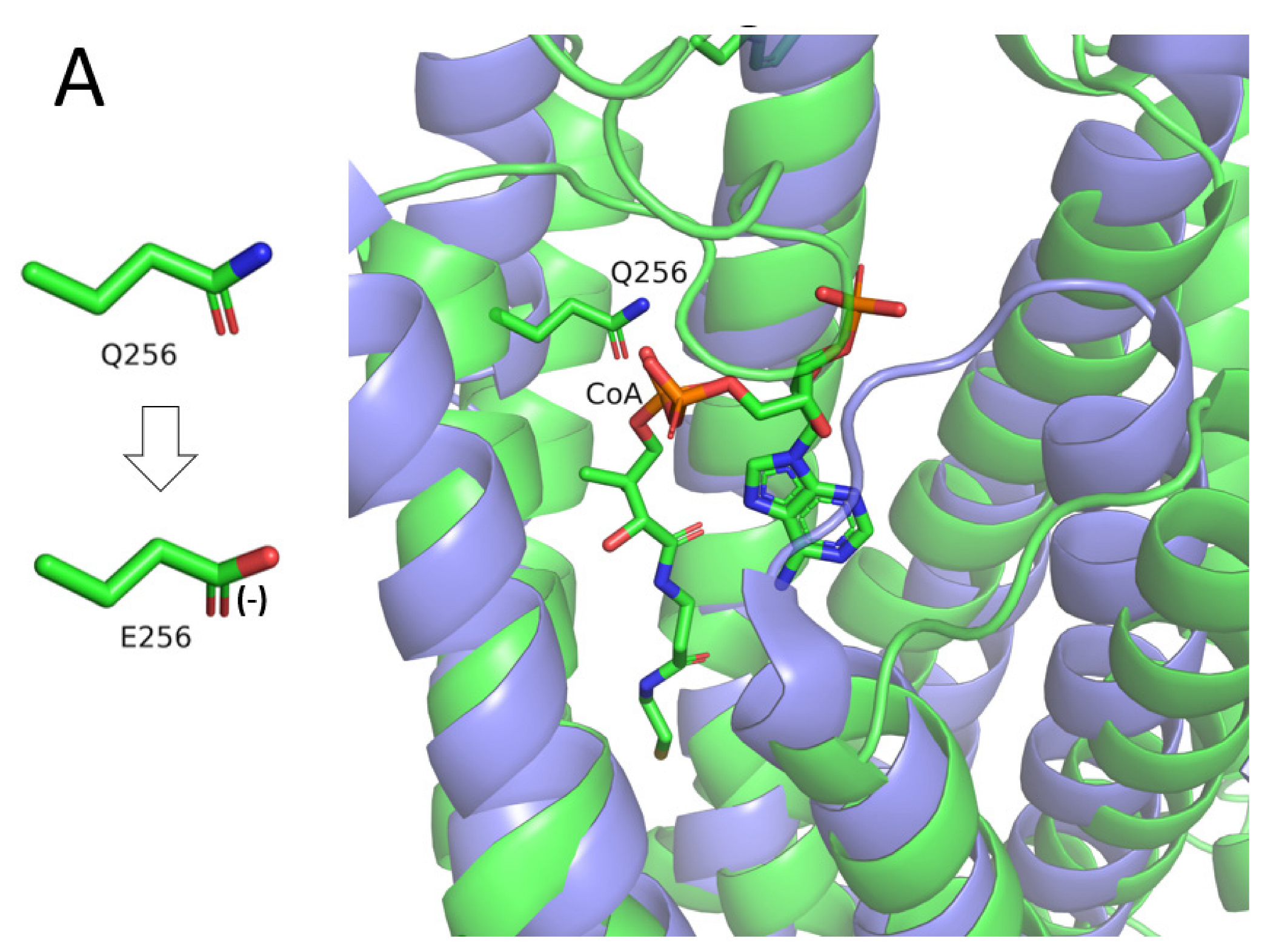
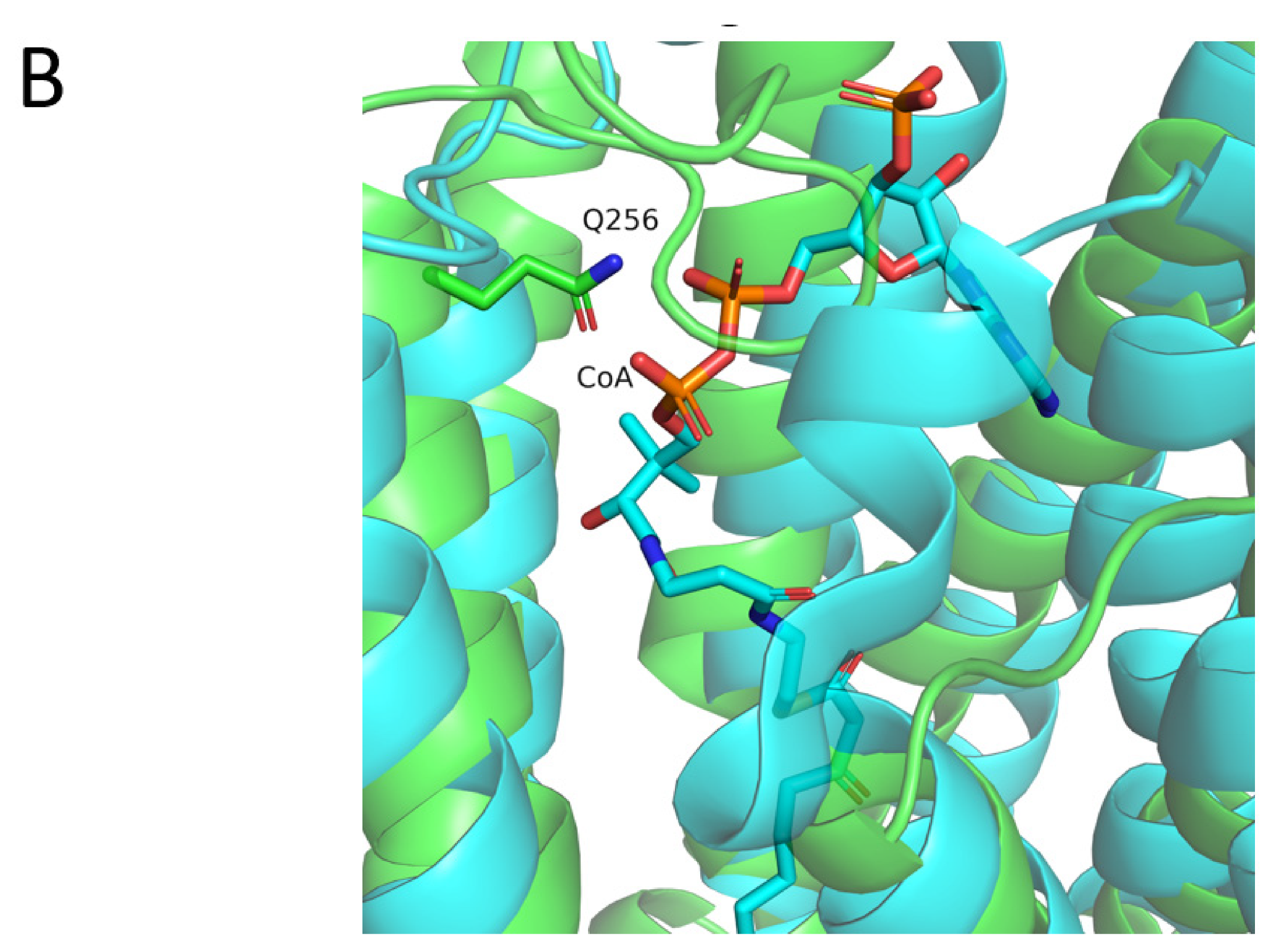
- I. Q256E (c.766C > G)
- The TACAN mechanosensitive ion channel, bound to coenzyme A (PDB ID: 7n0l, sequence identity to CLN8: 14%).
- ELOVL fatty acid elongase 7, bound to a coenzyme A-fatty acid conjugate (PDB ID: 6y7f, sequence identity to CLN8: 12%).
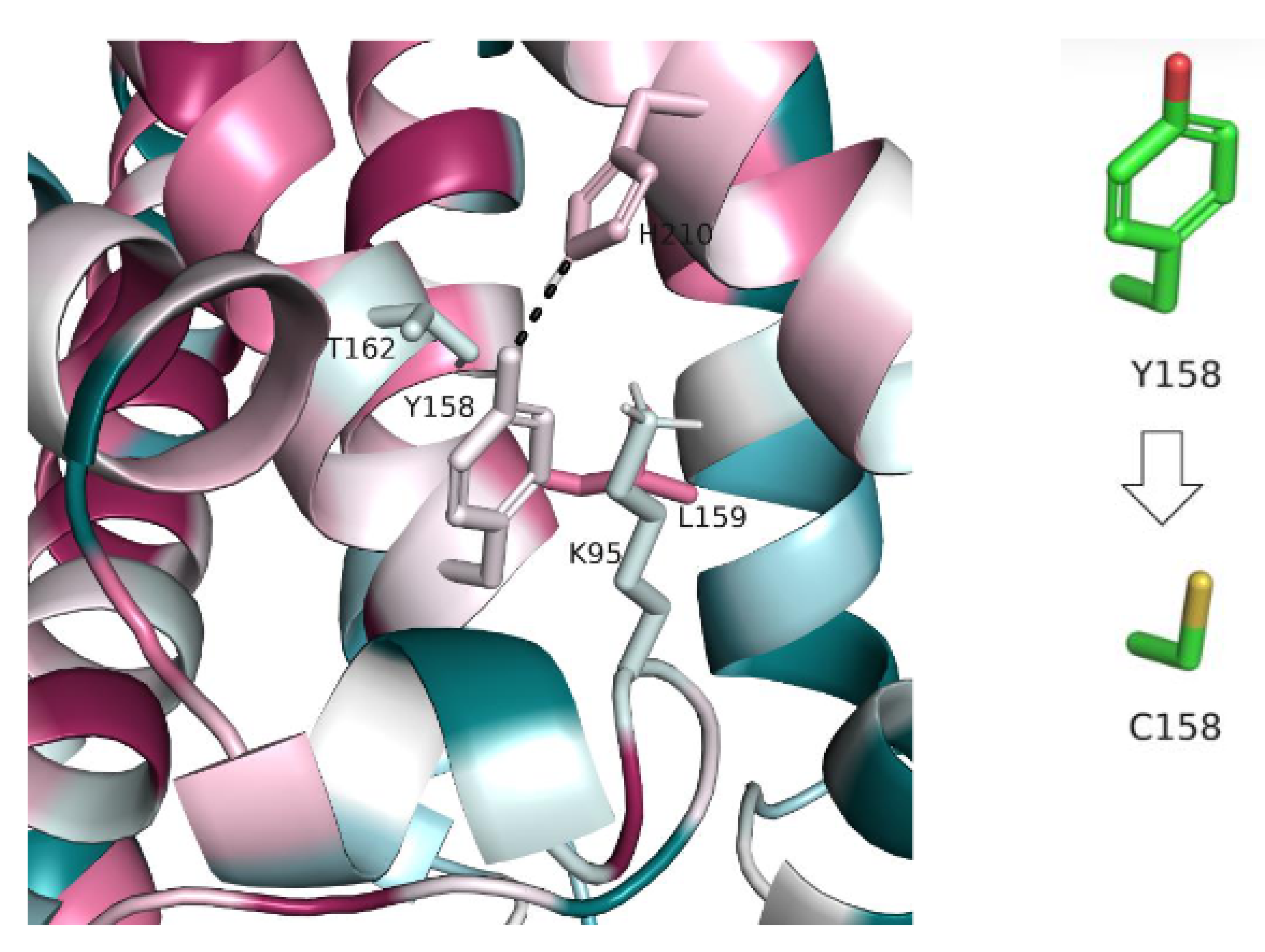
- II. Y158C (c.473A > G)
3.1.2. Mutations reported by Cannelli et al.’s Study in 2006
- I. Deletion After Position 22 (c.66delG)
- II. Y158C (c.473A > G)
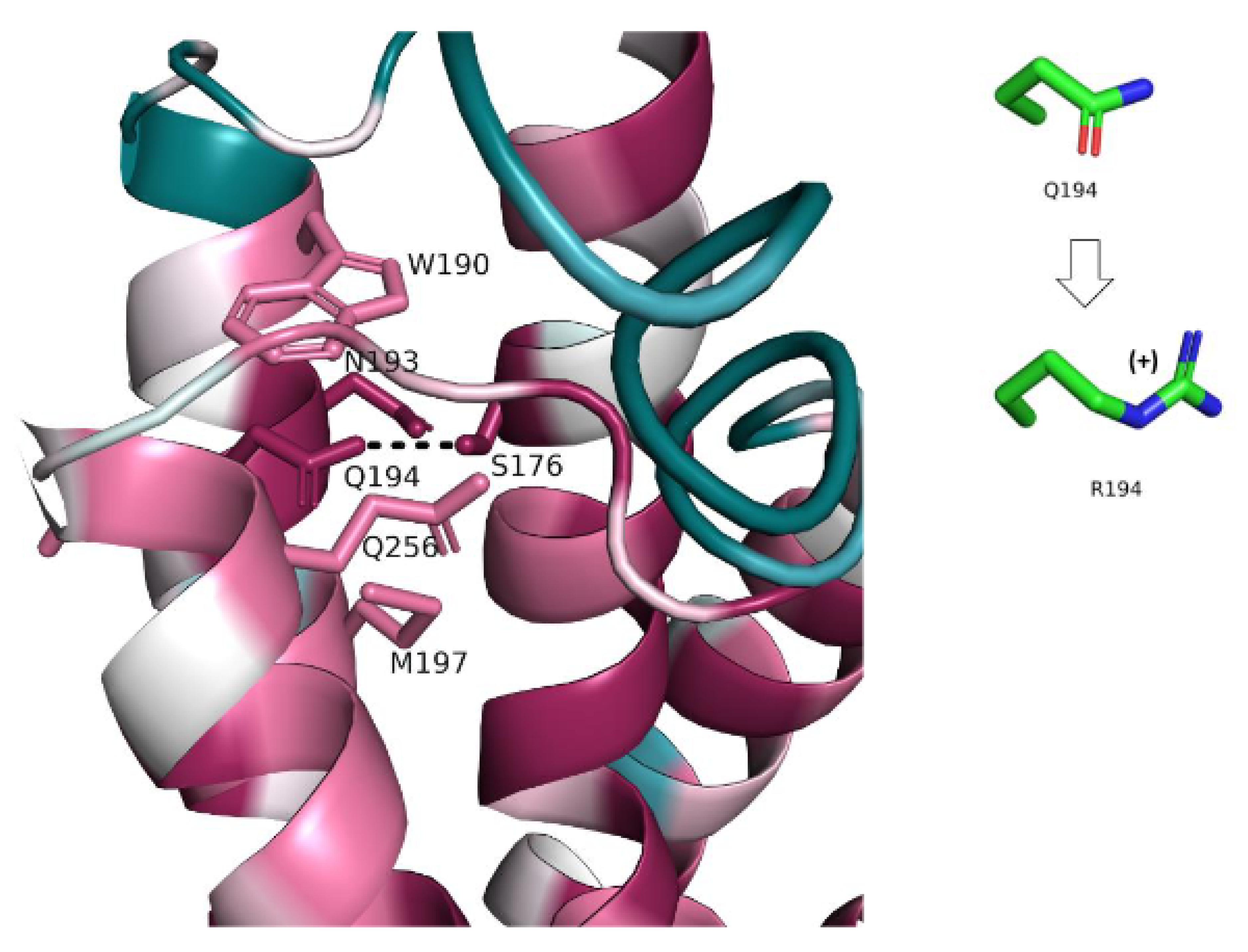
- III. Q194R (c.581A > G)
3.1.3. Mutation Reported by Allen et al.’s Study in 2012, Deletion after Position 188 (c.562_563delCT)
3.1.4. Mutations Reported by Beesley et al.’s Study in 2016
- I. Polypeptide Chain Termination After Position 255 (c.763C > T)
- II. L243P (c.728T > C)
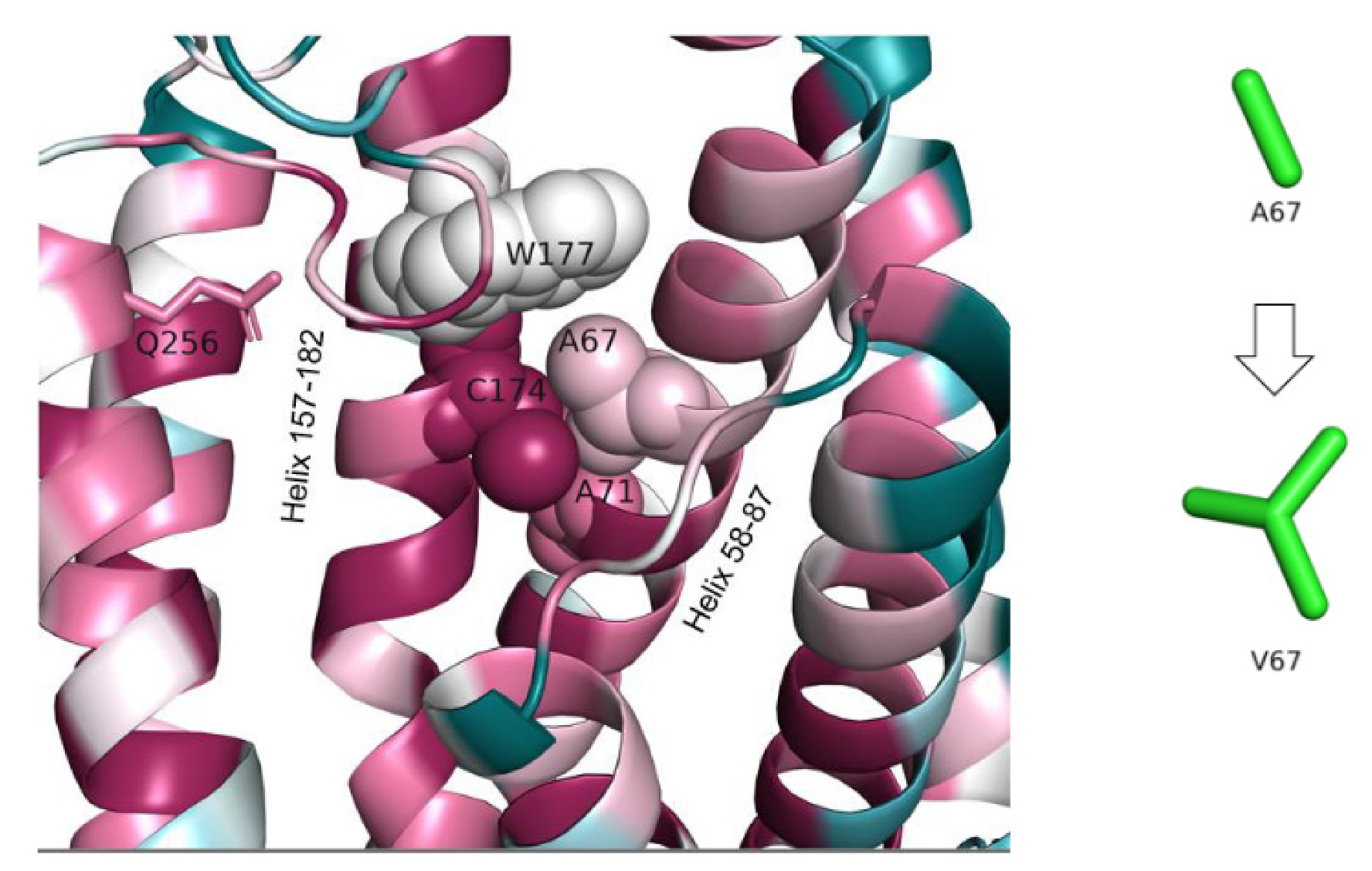
3.1.5. Mutation Reported by Sanchez et al.’s Study in 2016, A67V (c.200C > T)
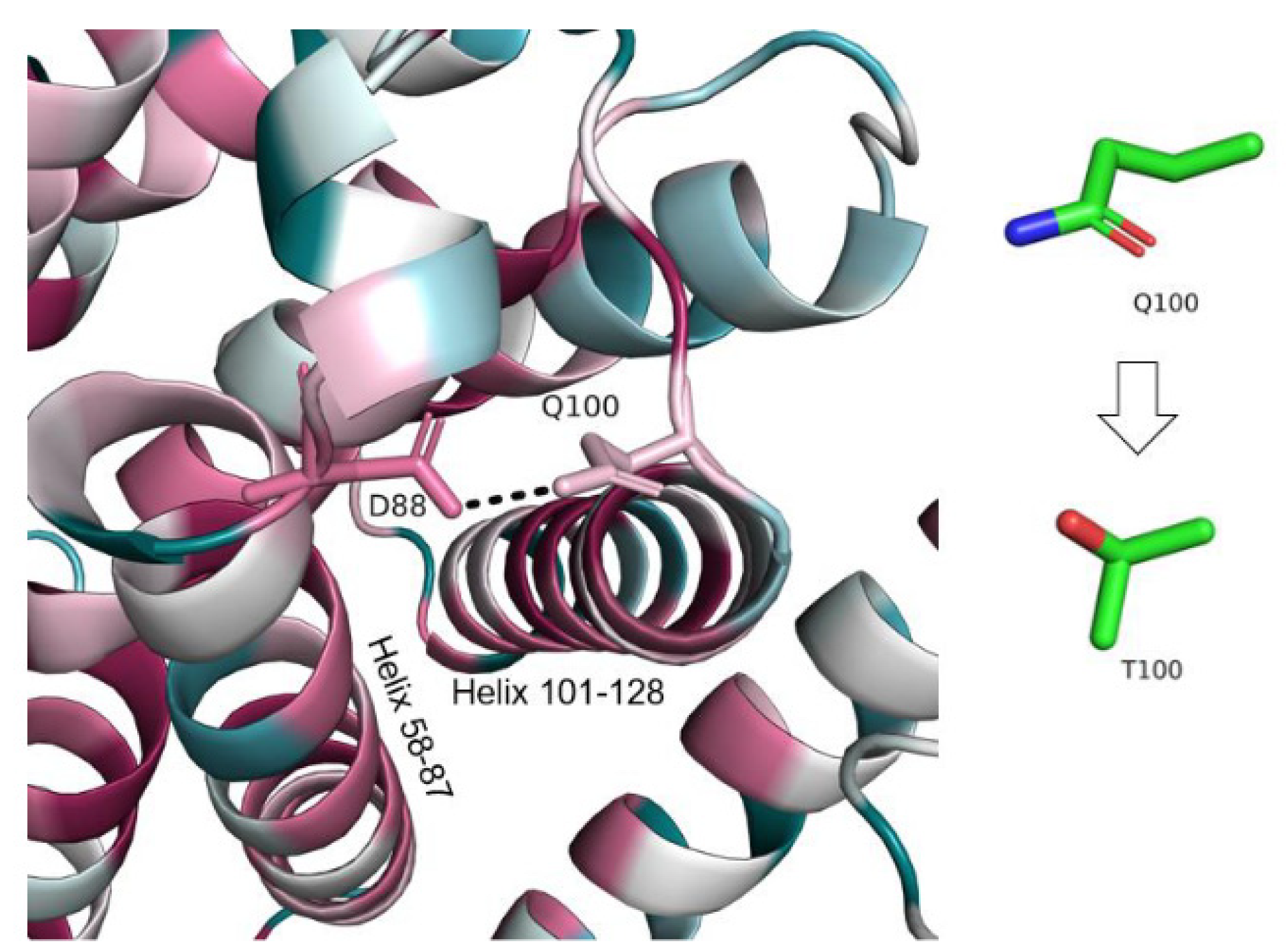
3.1.6. Mutation Reported by Gao et al.’s Study in 2018
- I. Q100T (c.298C > T)
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kim, W.D.; Wilson-Smillie, M.L.D.M.; Thanabalasingam, A.; Lefrancois, S.; Cotman, S.L.; Huber, R.J. Autophagy in the Neuronal Ceroid Lipofuscinoses (Batten Disease). Front. Cell Dev. Biol. 2022, 10, 812728. [Google Scholar] [CrossRef] [PubMed]
- Steinfeld, R.; Heim, P.; von Gregory, H.; Meyer, K.; Ullrich, K.; Goebel, H.H.; Kohlschütter, A. Late Infantile Neuronal Ceroid Lipofuscinosis: Quantitative Description of the Clinical Course in Patients with CLN2 Mutations. Am. J. Med. Genet. 2002, 112, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Warrier, V.; Vieira, M.; Mole, S.E. Genetic Basis and Phenotypic Correlations of the Neuronal Ceroid Lipofusinoses. Biochim. Biophys. Acta 2013, 1832, 1827–1830. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-L.; Zeng, Z.-Y.; Song, X.-W.; Hao, Z.-F.; Shi, Y.-W.; Tang, B.; Chen, S.-Q.; Gao, M.-M.; Di, W.; Long, Y.-S.; et al. A Novel CLN2/TPP1 Mutation in a Chinese Patient with Late Infantile Neuronal Ceroid Lipofuscinosis. Neurogenetics 2011, 12, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Huang, Y.; Meng, H.; Jiang, Y.; Wu, Y.; Xiong, H.; Wang, S.; Qin, J. Clinical Study in Chinese Patients with Late-Infantile Form Neuronal Ceroid Lipofuscinoses. Brain Dev. 2012, 34, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.E.; Cotman, S.L. Genetics of the Neuronal Ceroid Lipofuscinoses (Batten Disease). Biochim. Biophys. Acta 2015, 1852 Pt B, 2237–2241. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, J.B.; Chen, A.; Kaminsky, S.M.; Crystal, R.G.; Sondhi, D. Advances in the Treatment of Neuronal Ceroid Lipofuscinosis. Expert Opin. Orphan Drugs 2019, 7, 473–500. [Google Scholar] [CrossRef]
- Kohlschütter, A.; Schulz, A.; Bartsch, U.; Storch, S. Current and Emerging Treatment Strategies for Neuronal Ceroid Lipofuscinoses. CNS Drugs 2019, 33, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Hirvasniemi, A.; Lang, H.; Lehesjoki, A.E.; Leisti, J. Northern Epilepsy Syndrome: An Inherited Childhood Onset Epilepsy with Associated Mental Deterioration. J. Med. Genet. 1994, 31, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelnik, N.; Mahajna, M.; Iancu, T.C.; Sharony, R.; Zeigler, M. A Novel Mutation of the CLN8 Gene: Is There a Mediterranean Phenotype? Pediatr. Neurol. 2007, 36, 411–413. [Google Scholar] [CrossRef]
- Vantaggiato, C.; Redaelli, F.; Falcone, S.; Perrotta, C.; Tonelli, A.; Bondioni, S.; Morbin, M.; Riva, D.; Saletti, V.; Bonaglia, M.C.; et al. A Novel CLN8 Mutation in Late-Infantile-Onset Neuronal Ceroid Lipofuscinosis (LINCL) Reveals Aspects of CLN8 Neurobiological Function. Hum. Mutat. 2009, 30, 1104–1116. [Google Scholar] [CrossRef]
- Badura-Stronka, M.; Winczewska-Wiktor, A.; Pietrzak, A.; Hirschfeld, A.S.; Zemojtel, T.; Wołyńska, K.; Bednarek-Rajewska, K.; Seget-Dubaniewicz, M.; Matheisel, A.; Latos-Bielenska, A.; et al. CLN8 Mutations Presenting with a Phenotypic Continuum of Neuronal Ceroid Lipofuscinosis-Literature Review and Case Report. Genes 2021, 12, 956. [Google Scholar] [CrossRef]
- Kousi, M.; Lehesjoki, A.-E.; Mole, S.E. Update of the Mutation Spectrum and Clinical Correlations of over 360 Mutations in Eight Genes That Underlie the Neuronal Ceroid Lipofuscinoses. Hum. Mutat. 2012, 33, 42–63. [Google Scholar] [CrossRef]
- Jalanko, A.; Braulke, T. Neuronal Ceroid Lipofuscinoses. Biochim. Biophys. Acta 2009, 1793, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Winter, E.; Ponting, C.P. TRAM, LAG1 and CLN8: Members of a Novel Family of Lipid-Sensing Domains? Trends Biochem. Sci. 2002, 27, 381–383. [Google Scholar] [CrossRef]
- Mahajnah, M.; Zelnik, N. Phenotypic Heterogeneity in Consanguineous Patients with a Common CLN8 Mutation. Pediatr. Neurol. 2012, 47, 303–305. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly Accurate Protein Structure Prediction for the Human Proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. DUET: A Server for Predicting Effects of Mutations on Protein Stability Using an Integrated Computational Approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An Improved Methodology to Estimate and Visualize Evolutionary Conservation in Macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of Three-Dimensional Structural Information of Biological Macromolecules. Acta Crystallogr. D Biol. Crystallogr. 1998, 54 Pt 6 Pt 1, 1078–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, L. Using Dali for Protein Structure Comparison. Methods Mol. Biol. 2020, 2112, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, W.A.; Wheeler, R.B.; Sharp, J.D.; Bate, S.L.; Gardiner, R.M.; Ranta, U.S.; Lonka, L.; Williams, R.E.; Lehesjoki, A.E.; Mole, S.E. Turkish Variant Late Infantile Neuronal Ceroid Lipofuscinosis (CLN7) May Be Allelic to CLN8. Eur. J. Paediatr. Neurol. 2001, 5 (Suppl. A), 21–27. [Google Scholar] [CrossRef] [PubMed]
- Cannelli, N.; Cassandrini, D.; Bertini, E.; Striano, P.; Fusco, L.; Gaggero, R.; Specchio, N.; Biancheri, R.; Vigevano, F.; Bruno, C.; et al. Novel Mutations in CLN8 in Italian Variant Late Infantile Neuronal Ceroid Lipofuscinosis: Another Genetic Hit in the Mediterranean. Neurogenetics 2006, 7, 111–117. [Google Scholar] [CrossRef]
- Allen, N.M.; O’hIci, B.; Anderson, G.; Nestor, T.; Lynch, S.A.; King, M.D. Variant Late-Infantile Neuronal Ceroid Lipofuscinosis Due to a Novel Heterozygous CLN8 Mutation and de Novo 8p23.3 Deletion. Clin. Genet. 2012, 81, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Beesley, C.; Guerreiro, R.J.; Bras, J.T.; Williams, R.E.; Taratuto, A.L.; Eltze, C.; Mole, S.E. CLN8 Disease Caused by Large Genomic Deletions. Mol. Genet. Genom. Med. 2017, 5, 85–91. [Google Scholar] [CrossRef]
- Sanchez, R.L.; Yan, J.; Richards, S.; Mierau, G.; Wartchow, E.P.; Collins, C.D.; Shankar, S.P. Atypical Presentation of Neuronal Ceroid Lipofuscinosis Type 8 in a Sibling Pair and Review of the Eye Findings and Neurological Features. Am. J. Ophthalmol. Case Rep. 2016, 4, 50–53. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Xie, H.; Jiang, Q.; Wu, N.; Chen, X.; Chen, Q. Identification of Two Novel Null Variants in CLN8 by Targeted Next-Generation Sequencing: First Report of a Chinese Patient with Neuronal Ceroid Lipofuscinosis Due to CLN8 Variants. BMC Med. Genet. 2018, 19, 21. [Google Scholar] [CrossRef] [Green Version]
- Sharkia, R.; Khatib, M.; Sheikh-Muhammad, A.; Mahajnah, M.; Zalan, A. The Prevailing Trend of Consanguinity in the Arab Society of Israel: Is It Still a Challenge? J. Biosoc. Sci. 2021, 1–5. [Google Scholar] [CrossRef]
- Zalan, A. Consanguinity Status in the Arab Society of Israel: Is It Different? AJBSR 2021, 15, 66–76. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Study | Cannelli et al.’s Study in 2006 | Allen et al.’s Study in 2012 | Beesley et al.’s Study in 2016 | Sanchez et al.’s Study in 2016 | Gao et al.’s Study in2018 | Our Study |
|---|---|---|---|---|---|---|
| CLN8 Variant | ||||||
| Allele-1 | 1-c.66delG, p.(Gly22Serfs*5) 2-c.66delG, p.(Gly22Serfs*5) | c.562_563delCT, p.(Leu188Valfs*58) | 1–8p23.3 deletion, 54 Kb 2-c.763C > T, p.(Gln255*) | 1-c.200C > T (p.A67V) 2-c.200C > T (p.A67V) | 1-c.298C > T, p.(Gln100Ter) | 1-c.473A > G, p.(Tyr158Cys) 2-c.473A > G, p.(Tyr158Cys) |
| Allele-2 | 1-c.473A > G, p.(Tyr158Cys) 2-c.581A > G, p.(Gln194Arg) | 8p23.3 terminal deletion, de novo | 1-c.728T > C, p.(Leu243Pro) 2–8p23.3 deletion, 235 Kb | 1–8p23.3 deletion 2–8p23.3 deletion | 1-c.551G > A, p.(Trp184Ter) | 1-c.766C > G, p.(Gln256Glu) 2-c.766C > G, p.(Gln256Glu) |
| Clinical Features | ||||||
| Number of patients | 2 | 1 | 2 | 2 | 1 | 2 |
| Consanguinity | No | No | No | NM | No | No |
| Country of origin | Italy | Ireland | United Kingdom | USA | China | Israel |
| Gender | M/ M | M | F/M | F/M | M | F/F |
| Disease onset: (y) (A) Onset abnormal gait (y) | 3.5/4 3.5/4 | 4 4 | 4/ 3 (and 8m) 4 years 1 month/5 years and 2 months | 7/5 NM | 4 4 | 7.5/8 years 5 months No/12 years |
| (B) Onset visual impairment (y) | 3.5/NM | Present at 5 years, no record of when the onset was | NM/at onset leading to failure and retinal dystrophy | 14/5 | 7 | No/at 11 years limitation of smooth pursuit (oculomotor apraxia). No visual impairment |
| (C) Onset myoclonus (y) | 3.5/4 myoclonic jerks following febrile illness | Jerky ocular pursuit at 5 years no record of when the onset was | 4 years 4 months (myoclonic seizure)/at age 7 years had hyperkinetic limb movements | NM | NM | No/no |
| (D) Onset epilepsy (y) | 3/during 7 year follow up (myoclonic seizures) | 4.5 | 4 years 4 months / 4 years 9 months | 7/16 | 4 | 7.5/8 (and 5 m) |
| EEG | NM | Slow background, complex partial seizures | NM/abnormally slow background activity and runs of epileptiform discharges | NM | Irregular and slow background activity and high incidence of generalized sequences of atypical spike-wave discharges | Frontal sharp waves/normal |
| Loss of ambulation (y) | NM/during 7 year follow up | 5.5 | By 5 years 9 months/by 7 years | No/no | 8 years six months | No/no |
| Visual failure (y) | 3.5/during 7 year follow up | 5.5 | By 4 years 8 months/ 3 years 8 months | No/no | 8 years six months | No/no |
| Previous developmental delay | Yes (motor and speech)/yes (motor) | Yes Global (motor and speech) | Mild speech delay/ no | No/no | No | Mild learning disability ADHD/ADHD |
| Head circumference (centile) | NM | 50 cm (9th centile) | NM | NM | NM | 50th percentile/ 40th percentile |
| Psychomotor regression | 3.5/during 7 year follow up | Yes, at age 4 years | Yes, at onset/ yes, at 5 years and 2 months | No/no | Yes at 7 years old | Behavioral difficulties at age 11, speech disturbances at age 11 and 6 months/cognitive decline at 8 years 10 months |
| Brain imaging (MRI) | Cerebral and cerebellar atrophy/atrophied cerebellar vermis and cerebral cortex | Hyperintensity of PLIC, posterior dwm and centrum semi-oval (axial T2 FLAIR) plus cerebellar atrophy | Cerebellar atrophy, low signal change abnormality in thalami bilaterally/white matter abnormalities | NM | Diffuse cerebral and cerebellar atrophy | At age 8—mild cerebellar atrophy, MRI at age 12 years—worsening cerebellar atrophy/mild cerebellar atrophy, thalamic lesion |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharkia, R.; Zalan, A.; Zahalka, H.; Kessel, A.; Asaly, A.; Al-Shareef, W.; Mahajnah, M. CLN8 Gene Compound Heterozygous Variants: A New Case and Protein Bioinformatics Analyses. Genes 2022, 13, 1393. https://doi.org/10.3390/genes13081393
Sharkia R, Zalan A, Zahalka H, Kessel A, Asaly A, Al-Shareef W, Mahajnah M. CLN8 Gene Compound Heterozygous Variants: A New Case and Protein Bioinformatics Analyses. Genes. 2022; 13(8):1393. https://doi.org/10.3390/genes13081393
Chicago/Turabian StyleSharkia, Rajech, Abdelnaser Zalan, Hazar Zahalka, Amit Kessel, Ayman Asaly, Wasif Al-Shareef, and Muhammad Mahajnah. 2022. "CLN8 Gene Compound Heterozygous Variants: A New Case and Protein Bioinformatics Analyses" Genes 13, no. 8: 1393. https://doi.org/10.3390/genes13081393
APA StyleSharkia, R., Zalan, A., Zahalka, H., Kessel, A., Asaly, A., Al-Shareef, W., & Mahajnah, M. (2022). CLN8 Gene Compound Heterozygous Variants: A New Case and Protein Bioinformatics Analyses. Genes, 13(8), 1393. https://doi.org/10.3390/genes13081393