
LTBP4, SPP1, and CD40 Variants: Genetic Modifiers of Duchenne Muscular Dystrophy Analyzed in Serbian Patients

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Criteria
2.2. SNP Genotyping
2.3. Statistical Analysis
3. Results
3.1. Effect of Examined Individual Factors on LoA in DMD Patients
3.2. Cluster Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Emery, A.E.H. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Baxter, P. Treatment of the heart in Duchenne muscular dystrophy. Dev. Med. Child Neurol. 2006, 48, 163. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef] [Green Version]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018, 17, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Desguerre, I.; Christov, C.; Mayer, M.; Zeller, R.; Becane, H.M.; Bastuji-Garin, S.; Leturcq, F.; Chiron, C.; Chelly, J.; Gherardi, R.K. Clinical heterogeneity of duchenne muscular dystrophy (DMD): Definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS ONE 2009, 4, e4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbertclaude, V.; Hamroun, D.; Bezzou, K.; Bérard, C.; Boespflug-Tanguy, O.; Bommelaer, C.; Campana-Salort, E.; Cances, C.; Chabrol, B.; Commare, M.C.; et al. Motor and respiratory heterogeneity in Duchenne patients: Implication for clinical trials. Eur. J. Paediatr. Neurol. 2012, 16, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Hufton, M.; Roper, H. Variations in Duchenne muscular dystrophy course in a multi-ethnic UK population: Potential influence of socio-economic factors. Dev. Med. Child Neurol. 2017, 59, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Bello, L.; Kesari, A.; Gordish-Dressman, H.; Cnaan, A.; Morgenroth, L.P.; Punetha, J.; Duong, T.; Henricson, E.K.; Pegoraro, E.; McDonald, C.M.; et al. Genetic modifiers of ambulation in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study. Ann. Neurol. 2015, 77, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Bello, L.; Pegoraro, E. The “Usual Suspects”: Genes for Inflammation, Fibrosis, Regeneration, and Muscle Strength Modify Duchenne Muscular Dystrophy. J. Clin. Med. 2019, 8, 649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello, L.; D’Angelo, G.; Villa, M.; Fusto, A.; Vianello, S.; Merlo, B.; Sabbatini, D.; Barp, A.; Gandossini, S.; Magri, F.; et al. Genetic modifiers of respiratory function in Duchenne muscular dystrophy. Ann. Clin. Transl. Neurol. 2020, 7, 786–798. [Google Scholar] [CrossRef]
- Milic Rasic, V.; Vojinovic, D.; Pesovic, J.; Mijalkovic, G.; Lukic, V.; Mladenovic, J.; Kosac, A.; Novakovic, I.; Maksimovic, N.; Romac, S.; et al. Intellectual ability in the duchenne muscular dystrophy and dystrophin gene mutation location. Balkan J. Med. Genet. 2015, 17, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers. 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Manzur, A.Y.; Kuntzer, T.; Pike, M.; Swan, A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2016, 1, CD003725. [Google Scholar]
- Koeks, Z.; Bladen, C.L.; Salgado, D.; van Zwet, E.; Pogoryelova, O.; McMacken, G.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; et al. Clinical Outcomes in Duchenne Muscular Dystrophy: A Study of 5345 Patients from the TREAT-NMD DMD Global Database. J. Neuromuscul. Dis. 2017, 4, 293–306. [Google Scholar] [CrossRef] [Green Version]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Duong, T.; Joyce, N.C.; Hu, F.; Clemens, P.R.; Hoffman, E.P.; Cnaan, A.; Gordish-Dressman, H.; et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet 2018, 391, 451–461. [Google Scholar] [CrossRef]
- Herbelet, S.; De Paepe, B.; De Bleecker, J.L. Description of a Novel Mechanism Possibly Explaining the Antiproliferative Properties of Glucocorticoids in Duchenne Muscular Dystrophy Fibroblasts Based on Glucocorticoid Receptor GR and NFAT5. Int. J. Mol. Sci. 2020, 21, 9225. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Van Deutekom, J.C.; Fokkema, I.F.; Van Ommen, G.J.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef]
- Bello, L.; Morgenroth, L.P.; Gordish-Dressman, H.; Hoffman, E.P.; McDonald, C.M.; Cirak, S.; CINRG investigators. DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology 2016, 87, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Pegoraro, E.; Hoffman, E.P.; Piva, L.; Gavassini, B.F.; Cagnin, S.; Ermani, M.; Bello, L.; Soraru, G.; Pacchioni, B.; Bonifati, M.D.; et al. SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology 2011, 76, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagel, C.N.; Wasgewatte Wijesinghe, D.K.; Taghavi Esfandouni, N.; Mackie, E.J. Osteopontin, inflammation and myogenesis: Influencing regeneration, fibrosis and size of skeletal muscle. J. Cell Commun. Signal. 2014, 8, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramerova, I.; Kumagai-Cresse, C.; Ermolova, N.; Mokhonova, E.; Marinov, M.; Capote, J.; Becerra, D.; Quattrocelli, M.; Crosbie, R.H.; Welch, E.; et al. Spp1 (osteopontin) promotes TGFβ processing in fibroblasts of dystrophin-deficient muscles through matrix metalloproteinases. Hum. Mol. Genet. 2019, 28, 3431–3442. [Google Scholar] [CrossRef] [PubMed]
- Bello, L.; Piva, L.; Barp, A.; Taglia, A.; Picillo, E.; Vasco, G.; Pane, M.; Previtali, S.C.; Torrente, Y.; Gazzerro, E.; et al. Importance of SPP1 genotype as a covariate in clinical trials in Duchenne muscular dystrophy. Neurology 2012, 79, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Giacopelli, F.; Marciano, R.; Pistorio, A.; Catarsi, P.; Canini, S.; Karsenty, G.; Ravazzolo, R. Polymorphisms in the osteopontin promoter affect its transcriptional activity. Physiol. Genomics 2004, 20, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Piva, L.; Gavassini, B.F.; Bello, L.; Fanin, M.; Soraru, G.; Barp, A.; Ermani, M.; Angelini, C.; Ho_man, E.P.; Pegoraro, E. TGFBR2 but not SPP1 genotype modulates osteopontin expression in Duchenne muscular dystrophy muscle. J. Pathol. 2012, 228, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Flanigan, K.M.; Ceco, E.; Lamar, K.M.; Kaminoh, Y.; Dunn, D.M.; Mendell, J.R.; King, W.M.; Pestronk, A.; Florence, J.M.; Mathews, K.D.; et al. LTBP4 genotype predicts age of ambulatory loss in Duchenne muscular dystrophy. Ann. Neurol. 2013, 73, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Heydemann, A.; Ceco, E.; Lim, J.E.; Hadhazy, M.; Ryder, P.; Moran, J.L.; Beier, D.R.; Palmer, A.A.; McNally, E.M. Latent TGF-beta-binding protein 4 modifies muscular dystrophy in mice. J. Clin. Investig. 2009, 119, 3703–3712. [Google Scholar] [CrossRef] [Green Version]
- Lamar, K.-M.; Bogdanovich, S.; Gardner, B.B.; Gao, Q.Q.; Miller, T.; Earley, J.U.; Hadhazy, M.; Vo, A.H.; Wren, L.; Molkentin, J.D.; et al. Overexpression of latent TGF_ binding protein 4 in muscle ameliorates muscular dystrophy through myostatin and TGFβ. PLoS Genet. 2016, 12, e1006019. [Google Scholar] [CrossRef] [Green Version]
- Ceco, E.; Bogdanovich, S.; Gardner, B.; Miller, T.; DeJesus, A.; Earley, J.U.; Hadhazy, M.; Smith, L.R.; Barton, E.R.; Molkentin, J.D.; et al. Targeting latent TGFβ release in muscular dystrophy. Sci. Transl. Med. 2014, 6, 259. [Google Scholar] [CrossRef] [Green Version]
- Bello, L.; Flanigan, K.M.; Weiss, R.B.; Dunn, D.M.; Swoboda, K.J.; Gappmaier, E.; Howard, M.T.; Sampson, J.B.; Bromberg, M.B.; Butterfield, R.; et al. Association Study of Exon Variants in the NF-κB and TGFβ Pathways Identifies CD40 as a Modifier of Duchenne Muscular Dystrophy. Am. J. Hum. Genet. 2016, 99, 1163–1171. [Google Scholar] [CrossRef] [Green Version]
- Van den Bergen, J.C.; Ginjaar, H.B.; Niks, E.H.; Aartsma-Rus, A.; Verschuuren, J.J.G.M. Prolonged ambulation in Duchenne patients with a mutation amenable to exon 44 skipping. J. Neuromuscul. Dis. 2014, 1, 91–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winnard, A.V.; Mendell, J.R.; Prior, T.W.; Florence, J.; Burghes, A.H. Frameshift deletions of exons 3-7 and revertant fibers in Duchenne muscular dystrophy: Mechanisms of dystrophin production. Am. J. Hum. Genet. 1995, 56, 158–166. [Google Scholar] [PubMed]
- Wang, R.T.; Barthelemy, F.; Martin, A.S.; Douine, E.D.; Eskin, A.; Lucas, A.; Lavigne, J.; Peay, H.; Khanlou, N.; Sweeney, L.; et al. DMD genotype correlations from the Duchenne Registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum. Mutat. 2018, 39, 1193–1202. [Google Scholar] [CrossRef] [Green Version]
- Everitt, S.B.; Landau, S.; Leese, M.; Stahl, D. Cluster Analysis, 5th ed.; John Wiley & Sons Ltd.: Chichester, UK, 2011; pp. 73–110. [Google Scholar]
- Bzdok, D.; Altman, N.; Krzywinski, M. Statistics versus machine learning. Nat. Methods 2018, 15, 233–234. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 27 June 2022).
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Gualandi, F.; Rimessi, P.; Trabanelli, C.; Spitali, P.; Neri, M.; Patarnello, T.; Angelini, C.; Yau, S.C.; Abbs, S.; Muntoni, F.; et al. Intronic breakpoint definition and transcription analysis in DMD/BMD patients with deletion/duplication at the 5′ mutation hot spot of the dystrophin gene. Gene 2006, 370, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Doorenweerd, N.; Mahfouz, A.; van Putten, M.; Kaliyaperumal, R.; T’ Hoen, P.A.C.; Hendriksen, J.G.M.; Aartsma-Rus, A.M.; Verschuuren, J.J.G.M.; Niks, E.H.; Reinders, M.J.T.; et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci. Rep. 2017, 7, 12575. [Google Scholar] [CrossRef]
- Van den Bergen, J.C.; Hiller, M.; Böhringer, S.; Vijfhuizen, L.; Ginjaar, H.B.; Chaouch, A.; Bushby, K.; Straub, V.; Scoto, M.; Cirak, S.; et al. Validation of genetic modifiers for Duchenne muscular dystrophy: A multicentre study assessing SPP1 and LTBP4 variants. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Wang, L.; Li, Y.; Chen, Y.; Zhang, H.; Zhu, Y.; He, R.; Li, H.; Lin, J.; Zhang, Y.; et al. Genetic Modifiers of Duchenne Muscular Dystrophy in Chinese Patients. Front. Neurol. 2020, 11, 721. [Google Scholar] [CrossRef] [PubMed]
- Barp, A.; Bello, L.; Politano, L.; Melacini, P.; Calore, C.; Polo, A.; Vianello, S.; Sorarù, G.; Semplicini, C.; Pantic, B.; et al. Genetic Modifiers of Duchenne Muscular Dystrophy and Dilated Cardiomyopathy. PLoS ONE 2015, 10, e0141240. [Google Scholar] [CrossRef]
- Weiss, R.B.; Vieland, V.J.; Dunn, D.M.; Kaminoh, Y.; Flanigan, K.M.; United Dystrophinopathy Project. Long-range genomic regulators of THBS1 and LTBP4 modify disease severity in duchenne muscular dystrophy. Ann. Neurol. 2018, 84, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Quattrocelli, M.; Capote, J.; Ohiri, J.C.; Warner, J.L.; Vo, A.H.; Earley, J.U.; Hadhazy, M.; Demonbreun, A.R.; Spencer, M.J.; McNally, E.M. Genetic modifiers of muscular dystrophy act on sarcolemmal resealing and recovery from injury. PLoS Genet. 2017, 13, e1007070. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, M.W.; Houweling, P.J.; Thomas, K.C.; Gordish-Dressman, H.; Bello, L.; Cooperative International Neuromuscular Research Group (CINRG); Pegoraro, E.; Hoffman, E.P.; Head, S.I.; North, K.N. Evidence for ACTN3 as a genetic modifier of Duchenne muscular dystrophy. Nat Commun. 2017, 8, 14143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitali, P.; Zaharieva, I.; Bohringer, S.; Hiller, M.; Chaouch, A.; Roos, A.; Scotton, C.; Claustres, M.; Bello, L.; McDonald, C.M.; et al. TCTEX1D1 is a genetic modifier of disease progression in Duchenne muscular dystrophy. Eur. J. Hum. Genet. 2020, 28, 815–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
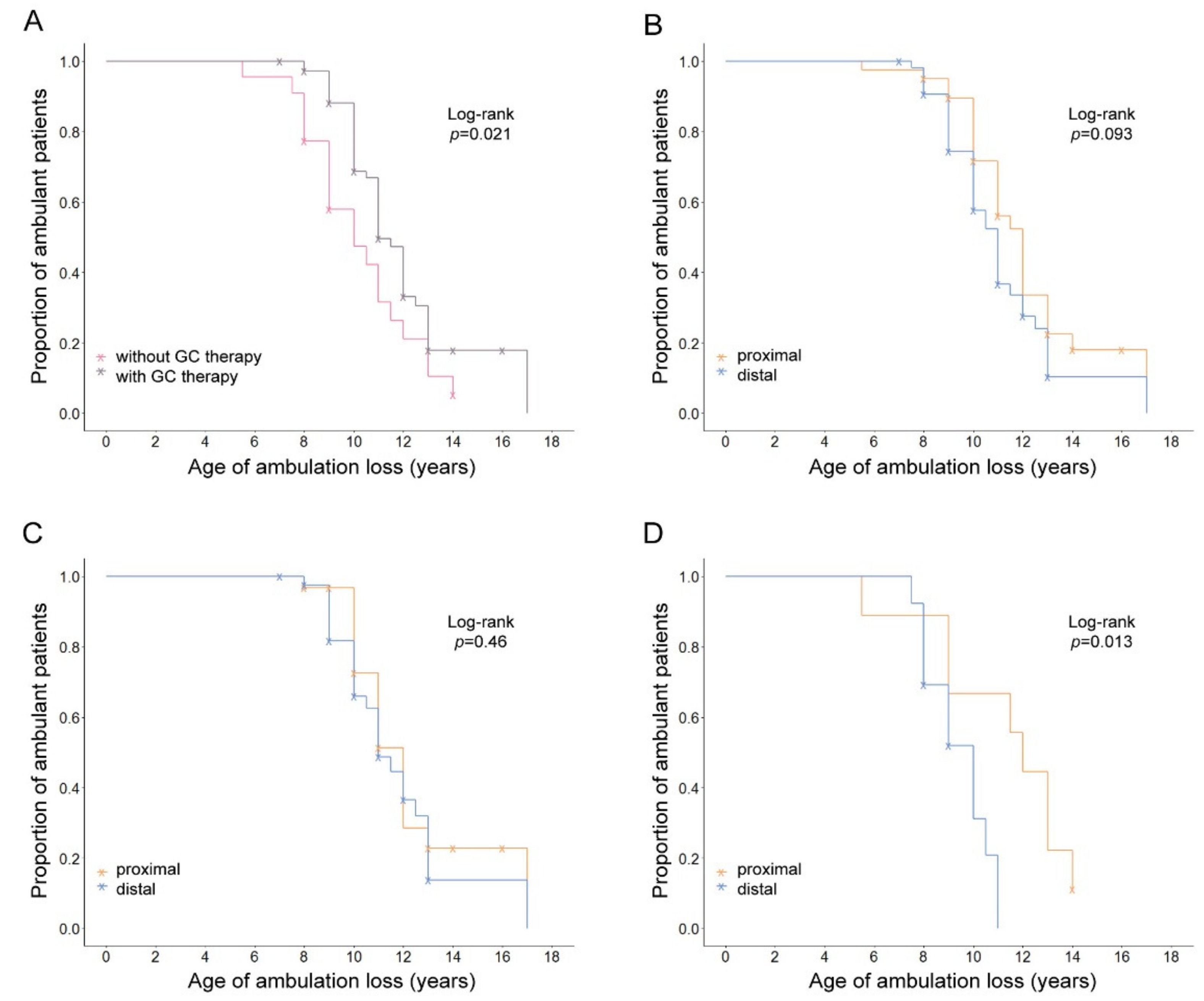

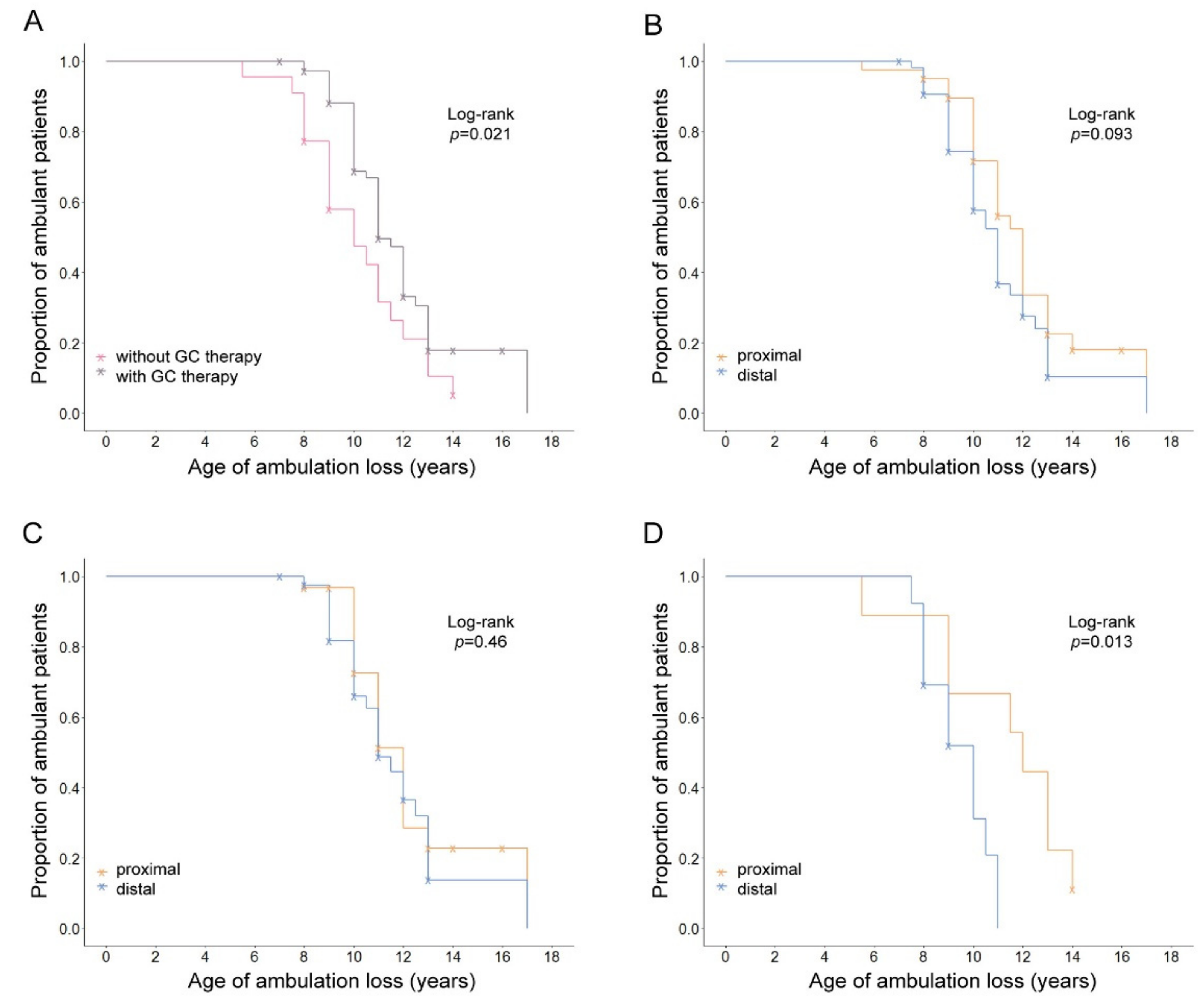{kind=link}
| GC Therapy | N (Events) | Mean Age of LoA Years (95% CI) | KM Log-Rank p | |
|---|---|---|---|---|
| Yes | 73 (45) | 11.14 (10.52–11.76) | 0.021 | |
| No | 22 (19) | 9.95 (8.9–11) | ||
| DMD mutation location | ||||
| Proximal | / | 40 (26) | 11.08 (10.21–11.95) | 0.093 |
| Distal | 55 (38) | 10.59 (9.88–11.3) | ||
| Proximal | Yes | 31 (18) | 11.17 (10.25–12.09) | 0.46 |
| Distal | 42 (27) | 11.13 (10.24–12.02) | ||
| Proximal | No | 9 (8) | 10.88 (8.51–13.25) | 0.013 |
| Distal | 13 (11) | 9.27 (8.4–10.14) | ||
| SPP1 (rs28357094) | 93 (62) | |||
| TT | / | 55 (36) | 10.94 (10.23–11.65) | 0.32 |
| TG + GG | 38 (26) | 10.62 (9.68–11.56) | ||
| TT | Yes | 46 (28) | 11.02 (10.19–11.85) | 0.79 |
| TG + GG | 25 (15) | 11.5 (10.35–12.65) | ||
| TT | No | 9 (8) | 10.69 (8.95–12.43) | 0.69 |
| TG + GG | 13 (11) | 9.41 (7.95–10.87) | ||
| CD40 (rs1883832) | 95 (64) | |||
| CC | / | 40 (31) | 10.69 (9.94–11.44) | 0.24 |
| CT + TT | 55 (33) | 10.88 (10.07–11.69) | ||
| CC | Yes | 27 (20) | 10.95 (9.99–11.91) | 0.45 |
| CT + TT | 46 (25) | 11.3 (10.42–12.18) | ||
| CC | No | 13 (11) | 10.23 (8.83–11.63) | 0.8 |
| CT + TT | 9 (8) | 9.56 (7.57–11.55) | ||
| LTBP4 (Haplotype) | 89 (58) | |||
| Other/other | / | 41 (27) | 10.39 (9.5–11.28) | 0.61 |
| Other/IAAM + IAAM/IAAM | 48 (31) | 10.69 (10.12–11.26) | ||
| Other/other | Yes | 34 (22) | 11.02 (10.16–11.88) | 0.43 |
| Other/IAAM + IAAM/IAAM | 33 (17) | 10.62 (9.9–11.34) | ||
| Other/other | No | 7 (5) | 7.6 (5.99–9.21) | 0.43 |
| Other/IAAM + IAAM/IAAM | 15 (14) | 10.79 (9.77–11.81) | ||
| Other/other | / | 41 (27) | 10.39 (9.5–11.28) | 0.56 |
| Other/IAAM | 40 (26) | 10.54 (9.91–11.17) | ||
| IAAM/IAAM | 8 (5) | 11.5 (9.74–13.26) | ||
| Other/other | Yes | 34 (22) | 11.02 (10.16–11.88) | 0.62 |
| Other/IAAM | 28 (15) | 10.63 (9.8–11.46) | ||
| IAAM/IAAM | 5 (2) | 10.5 (4.15–16.85) | ||
| Other/other | No | 7 (5) | 7.6 (5.99–9.21) | 0.54 |
| Other/IAAM | 12 (11) | 10.41 (9.26–11.56) | ||
| IAAM/IAAM | 3 (3) | 12.17 (8.58–15.76) | ||
| Other/other + other/IAAM | / | 81 (53) | 10.46 (9.93–10.99) | 0.29 |
| IAAM/IAAM | 8 (5) | 11.5 (9.74–13.26) | ||
| Other/other + other/IAAM | Yes | 62 (37) | 10.86 (10.27–11.45) | 0.43 |
| IAAM/IAAM | 5 (2) | 10.5 (4.15–16.85) | ||
| Other/other + other/IAAM | No | 19 (16) | 9.53 (8.44–10.62) | 0.34 |
| IAAM/IAAM | 3 (3) | 12.17 (8.58–15.76) |
| N (Events) | HR (95% CI) | Z-Score | p-Value | Cox p-Value | |
|---|---|---|---|---|---|
| GC therapy | 0.068 | ||||
| Nontreated | 22 (19) | 1 | 0.01 | ||
| Treated | 73 (45) | 0.44 (0.23–0.83) | −2.51 | ||
| DMD mutation location | |||||
| Proximal | 40 (26) | 1 | 0.03 | ||
| Distal | 55 (38) | 1.92 (1.07–3.47) | 2.18 | ||
| SPP1 (rs28357094) | 93 (62) | ||||
| TT | 55 (36) | 1 | 0.9 | ||
| TG + GG | 38 (26) | 1.03 (0.60–1.78) | 0.12 | ||
| CD40 (rs1883832) | 95 (64) | ||||
| CC | 40 (31) | 1 | 0.59 | ||
| CT + TT | 55 (33) | 0.86 (0.5–1.48) | −0.54 | ||
| LTBP4 (Haplotype) | 89 (58) | ||||
| Other/other | 41 (27) | 1 | 0.25 | ||
| Other/IAAM + IAAM/IAAM | 48 (31) | 0.72 (0.41–1.26) | −1.15 | ||
| Interaction | |||||
| GC treatment × DMD mutation (distal) | 42 (27) | 0.21 (0.06–0.74) | −2.42 | 0.02 | 0.056 |
| GC treatment × SPP1 rs28357094 (TG + GG) | 25 (15) | 0.69 (0.19–2.55) | −0.56 | 0.58 | |
| GC treatment × CD40 rs1883832 (CT + TT) | 46 (25) | 0.68 (0.19–2.38) | −0.6 | 0.55 | |
| GC treatment × LTBP4 (other/IAAM + IAAM/IAAM) | 33 (17) | 1.25 (0.29–5.4) | 0.3 | 0.76 |
| Variable | Description | Cluster I | Cluster II | p-Value |
|---|---|---|---|---|
| N = 41 | N = 23 | |||
| SPP1 rs28357094 * | TT | 24 | 14 | 1 ** |
| TG + GG | 17 | 9 | ||
| CD40 rs1883832 * | CC | 13 | 18 | 5.7 × 10−4 ** |
| CT + TT | 28 | 5 | ||
| LTBP4 haplotypes * | Other/other | 26 | 1 | 2.8 × 10−6 ** |
| Other/IAAM + IAAM/IAAM | 15 | 22 | ||
| Location of mutation | Proximal | 12 | 14 | 0.018 ** |
| Distal | 29 | 9 | ||
| Age at LoA | ≤10.75 years | 24 | 8 | 0.06 *** |
| >10.75 years | 17 | 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosac, A.; Pesovic, J.; Radenkovic, L.; Brkusanin, M.; Radovanovic, N.; Djurisic, M.; Radivojevic, D.; Mladenovic, J.; Ostojic, S.; Kovacevic, G.; et al. LTBP4, SPP1, and CD40 Variants: Genetic Modifiers of Duchenne Muscular Dystrophy Analyzed in Serbian Patients. Genes 2022, 13, 1385. https://doi.org/10.3390/genes13081385
Kosac A, Pesovic J, Radenkovic L, Brkusanin M, Radovanovic N, Djurisic M, Radivojevic D, Mladenovic J, Ostojic S, Kovacevic G, et al. LTBP4, SPP1, and CD40 Variants: Genetic Modifiers of Duchenne Muscular Dystrophy Analyzed in Serbian Patients. Genes. 2022; 13(8):1385. https://doi.org/10.3390/genes13081385
Chicago/Turabian StyleKosac, Ana, Jovan Pesovic, Lana Radenkovic, Milos Brkusanin, Nemanja Radovanovic, Marina Djurisic, Danijela Radivojevic, Jelena Mladenovic, Slavica Ostojic, Gordana Kovacevic, and et al. 2022. "LTBP4, SPP1, and CD40 Variants: Genetic Modifiers of Duchenne Muscular Dystrophy Analyzed in Serbian Patients" Genes 13, no. 8: 1385. https://doi.org/10.3390/genes13081385
APA StyleKosac, A., Pesovic, J., Radenkovic, L., Brkusanin, M., Radovanovic, N., Djurisic, M., Radivojevic, D., Mladenovic, J., Ostojic, S., Kovacevic, G., Kravljanac, R., Savic Pavicevic, D., & Milic Rasic, V. (2022). LTBP4, SPP1, and CD40 Variants: Genetic Modifiers of Duchenne Muscular Dystrophy Analyzed in Serbian Patients. Genes, 13(8), 1385. https://doi.org/10.3390/genes13081385