
Visual Clustering of Transcriptomic Data from Primary and Metastatic Tumors—Dependencies and Novel Pitfalls

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Bioinformatic Analysis
3. Results
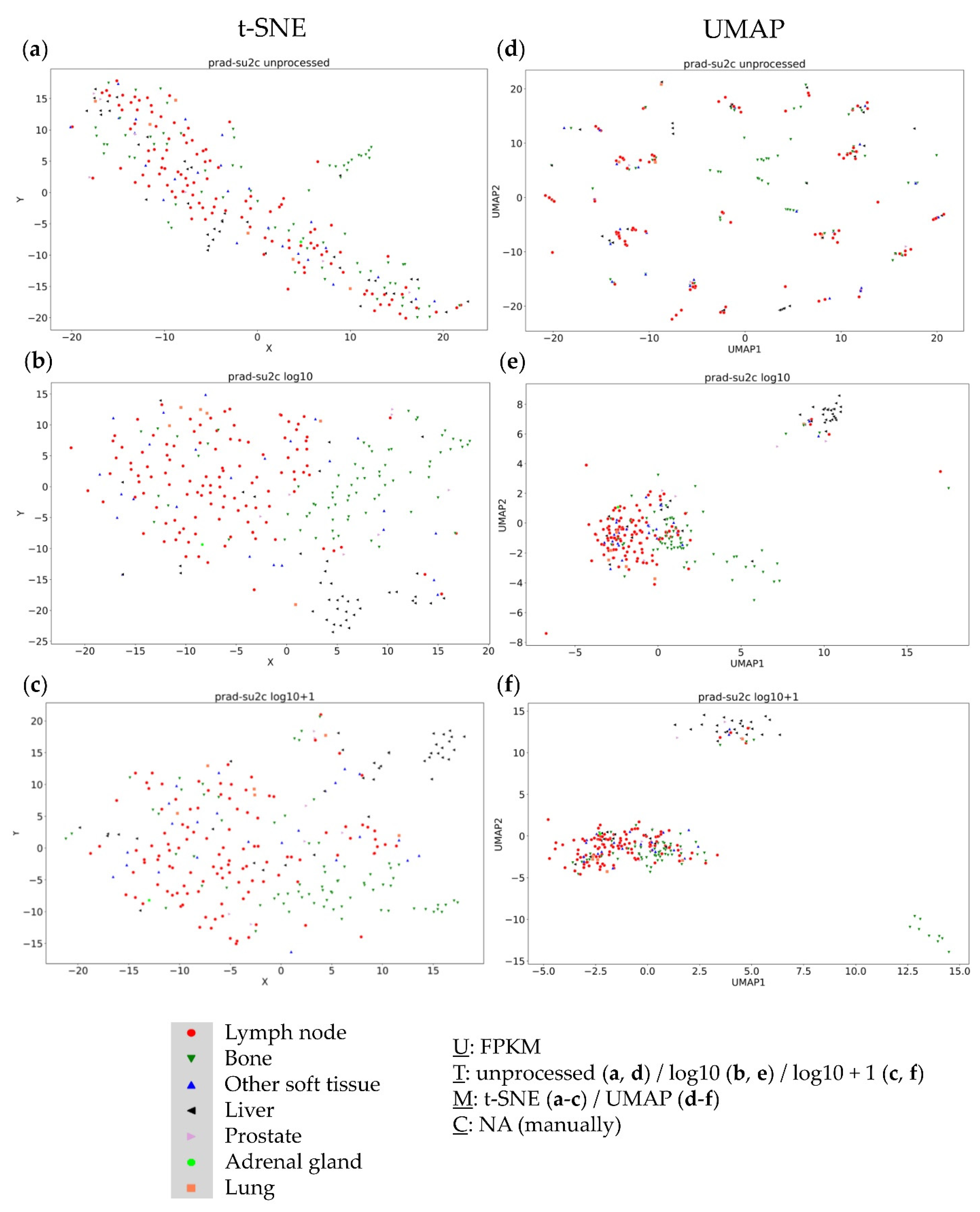
3.1. Analysis of the PRAD-SU2C (Dream Team) Dataset
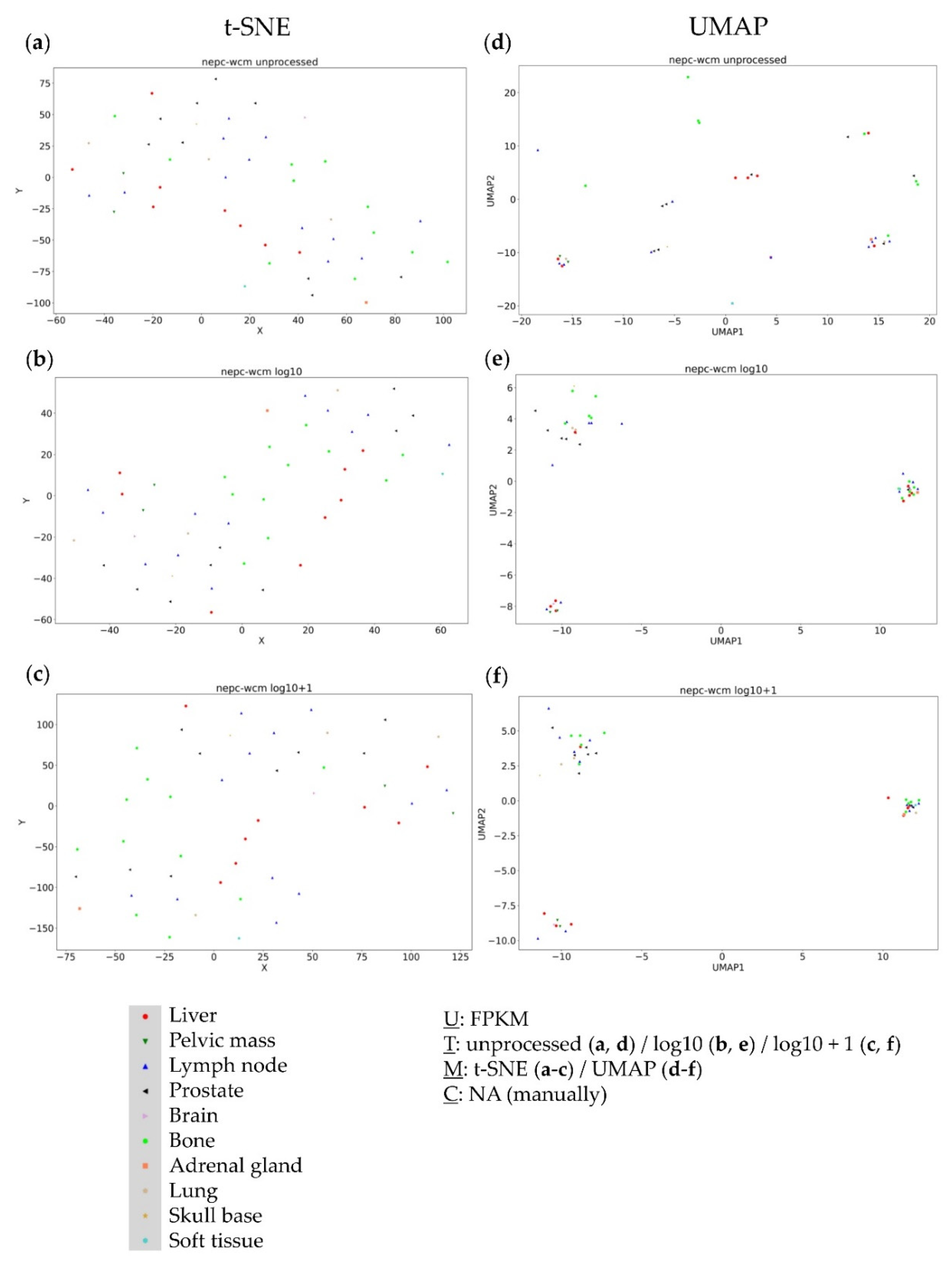
3.2. Analysis of the NEPC WCM (Neuroendocrine Prostate Cancer) Dataset
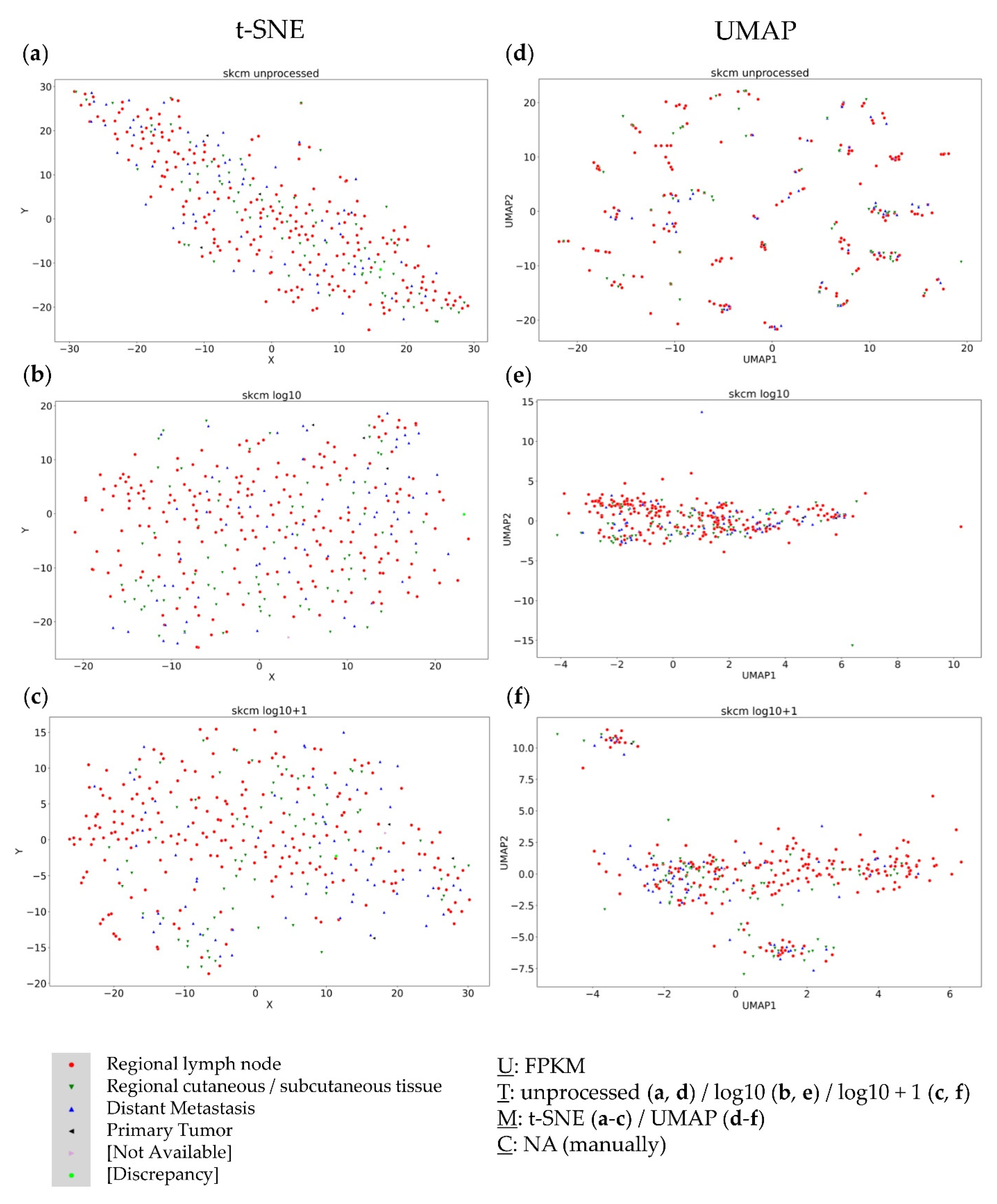
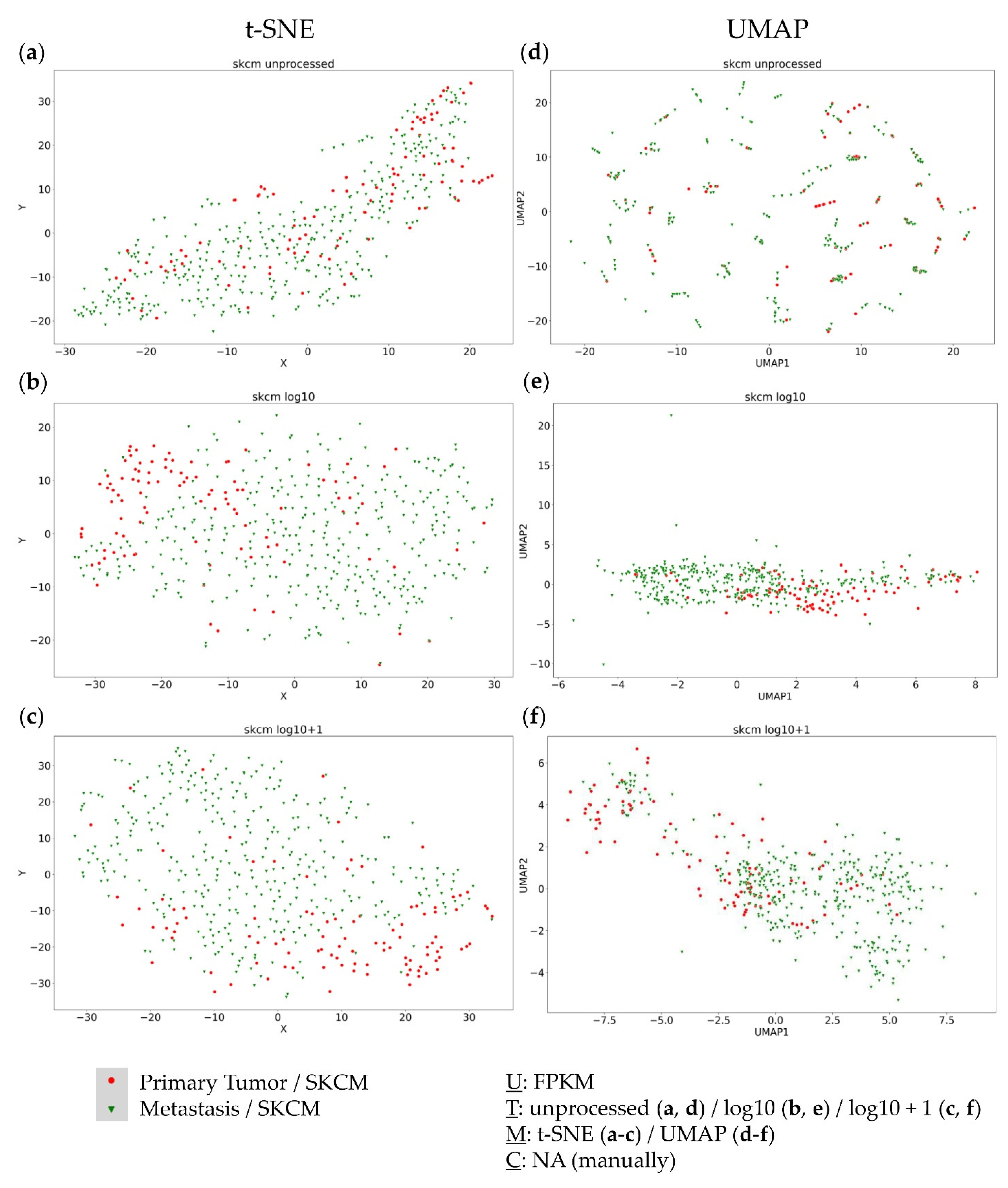
3.3. Analysis of the Metastatic Samples of TCGA-SKCM Dataset
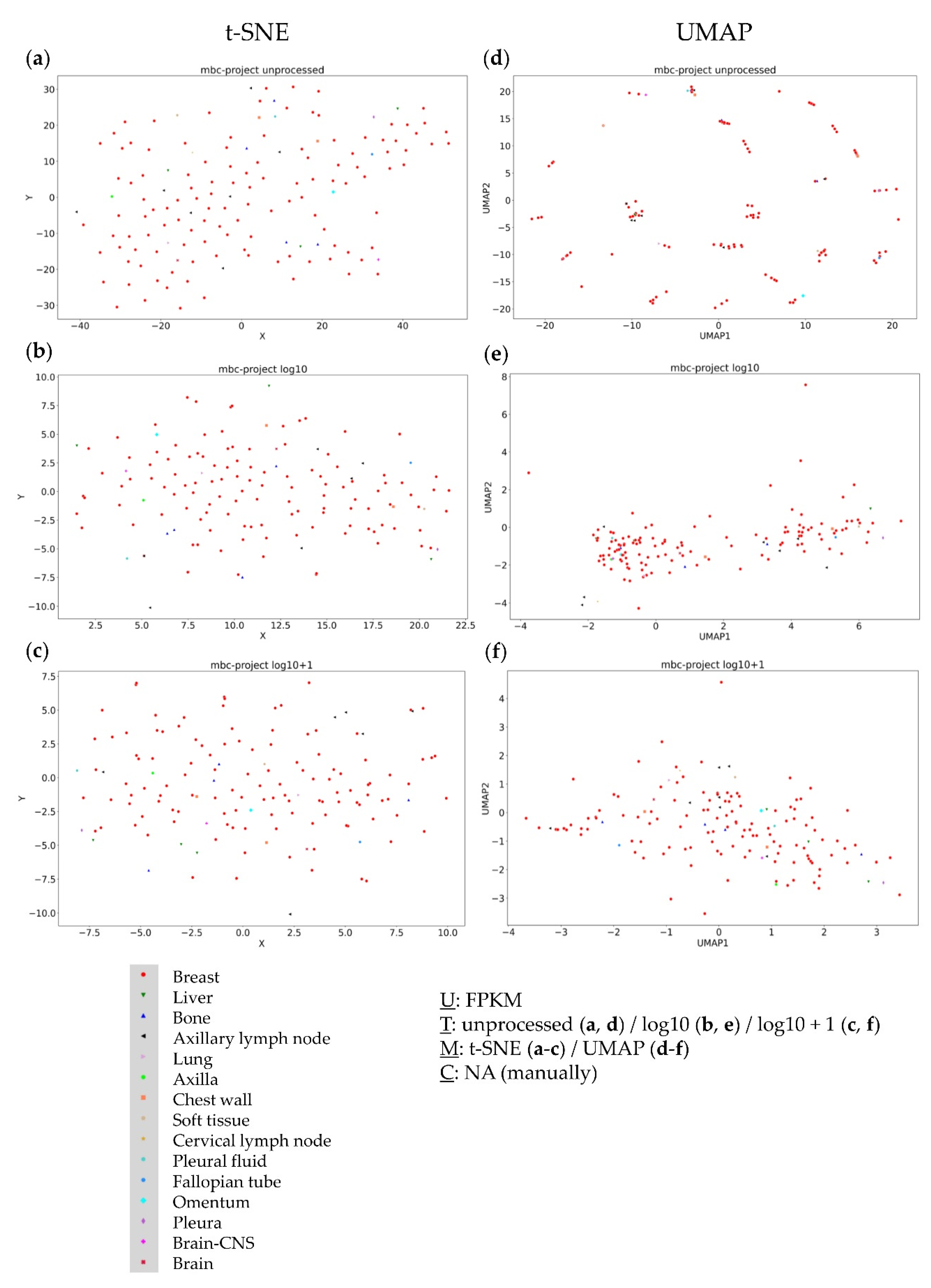
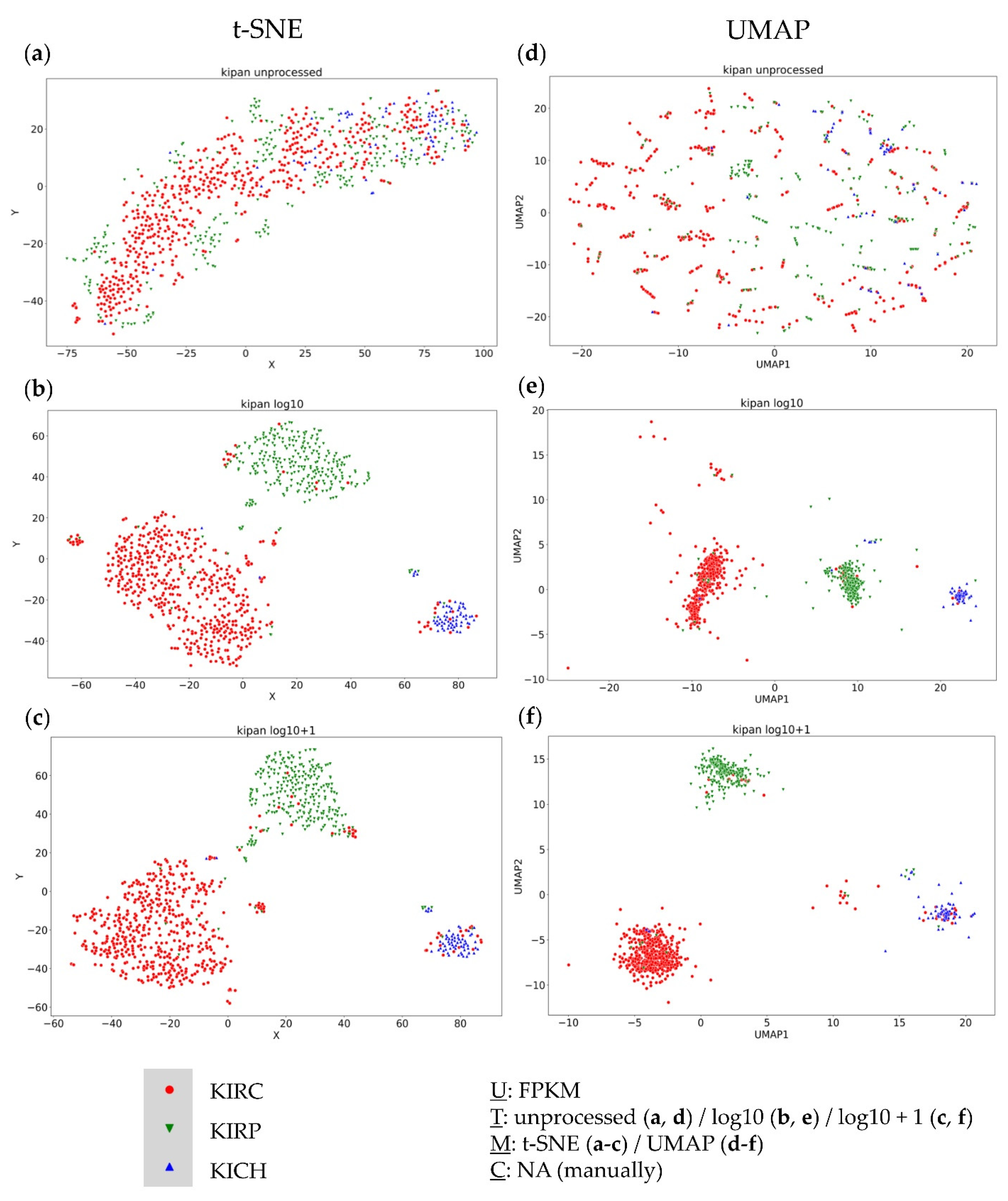
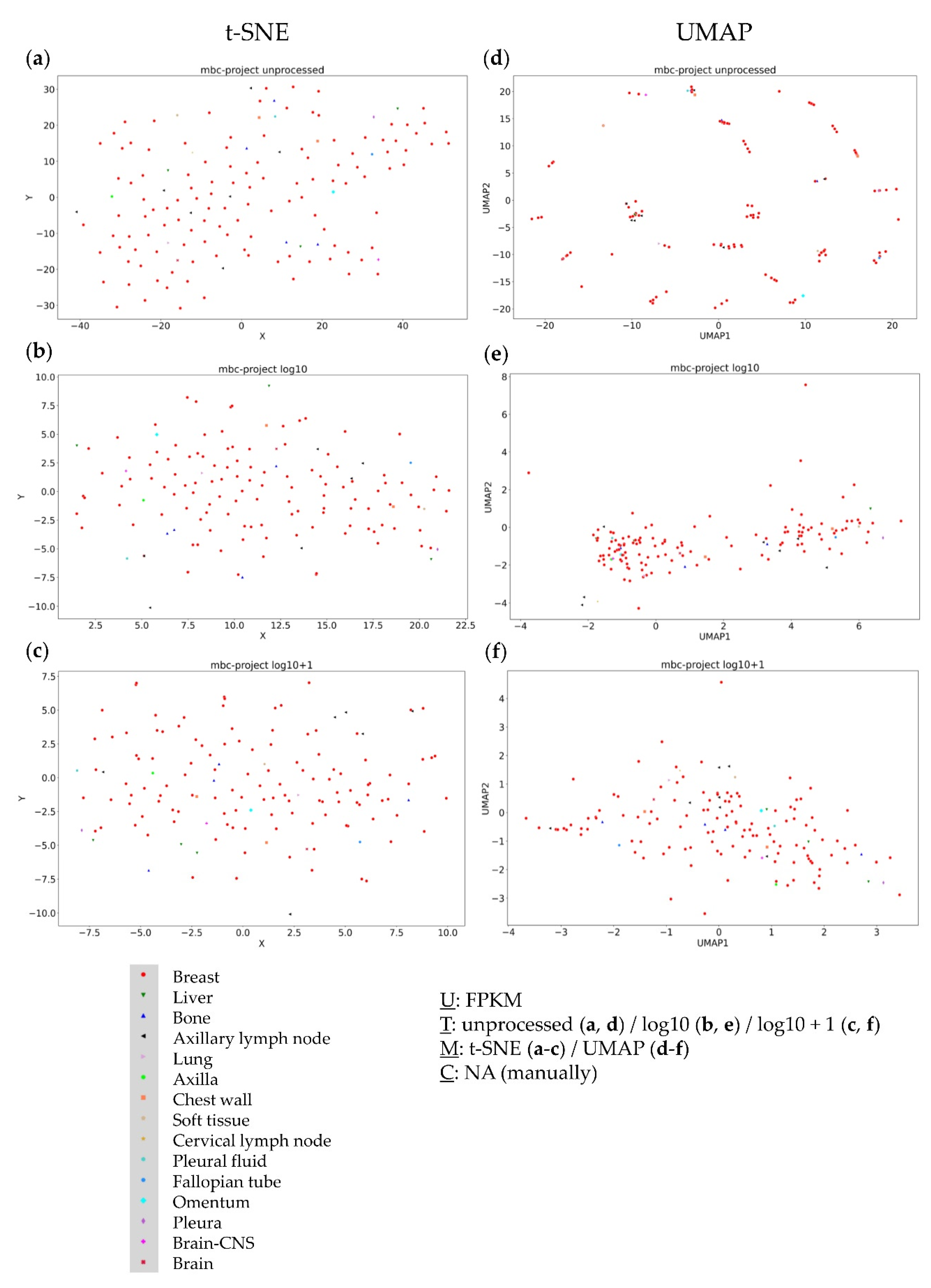
3.4. Further Evaluation of Cluster Formation Based on Data Dimension Reduction Methods and Data Transformations
3.5. Combined Analysis of Primary and Metastatic Samples of the Same Entity
4. Discussion
4.1. The Impact of Data Transformation on Cluster Formation within Data Dimension Reduction
4.2. Primary Tumors and Metastases of the Same Entity Share Common Transcriptomic Features
4.3. Addressing Pitfalls in Visual Clustering
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, Q.; Li, J.; Zhu, S.; Wu, J.; Chen, C.; Liu, Q.; Wei, W.; Zhang, Y.; Sun, S. Breast cancer subtypes predict the preferential site of distant metastases: A SEER based study. Oncotarget 2017, 8, 27990–27996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhang, R.; Michalski, C.W.; Liu, B.; Liao, Q.; Kleeff, J. Surgery for synchronous and metachronous single-organ metastasis of pancreatic cancer: A SEER database analysis and systematic literature review. Sci. Rep. 2020, 10, 4444. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.M.; Truty, M.J.; Nogueras-Gonzalez, G.M.; Fleming, J.B.; Vauthey, J.-N.; Pisters, P.W.T.; Lee, J.E.; Rice, D.C.; Hofstetter, W.L.; Wolff, R.A.; et al. Selective reoperation for locally recurrent or metastatic pancreatic ductal adenocarcinoma following primary pancreatic resection. J. Gastrointest. Surg. 2012, 16, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Nishizaki, T.; DeVries, S.; Chew, K.; Goodson, W.H.; Ljung, B.-M.; Thor, A.; Waldman, F.M. Genetic alterations in primary breast cancers and their metastases: Direct comparison using modified comparative genomic hybridization. Genes Chromosom. Cancer 1997, 19, 267–272. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [Green Version]
- Siraj, S.; Masoodi, T.; Siraj, A.K.; Azam, S.; Qadri, Z.; Ahmed, S.O.; AlBalawy, W.N.; Al-Obaisi, K.A.; Parvathareddy, S.K.; AlManea, H.M.; et al. Clonal Evolution and Timing of Metastatic Colorectal Cancer. Cancers 2020, 12, 2938. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Esmaeli, B. Patterns of regional and distant metastasis in patients with conjunctival melanoma Experience at a cancer center over four decades. Ophthalmology 2001, 108, 2101–2105. [Google Scholar] [CrossRef]
- Cairns, J. Mutation selection and the natural history of cancer. Nature 1975, 255, 197–200. [Google Scholar] [CrossRef]
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef]
- Klein, A.; Olendrowitz, C.; Schmutzler, R.; Hampl, J.; Schlag, P.M.; Maass, N.; Arnold, N.; Wessel, R.; Ramser, J.; Meindl, A.; et al. Identification of brain- and bone-specific breast cancer metastasis genes. Cancer Lett. 2009, 276, 212–220. [Google Scholar] [CrossRef]
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014, 15, 454. [Google Scholar] [CrossRef]
- Goswami, R.S.; Patel, K.P.; Singh, R.R.; Meric-Bernstam, F.; Kopetz, E.S.; Subbiah, V.; Alvarez, R.H.; Davies, M.A.; Jabbar, K.J.; Roy-Chowdhuri, S.; et al. Hotspot mutation panel testing reveals clonal evolution in a study of 265 paired primary and metastatic tumors. Clin. Cancer Res. 2015, 21, 2644–2651. [Google Scholar] [CrossRef] [Green Version]
- Vignot, S.; Lefebvre, C.; Frampton, G.M.; Meurice, G.; Yelensky, R.; Palmer, G.; Capron, F.; Lazar, V.; Hannoun, L.; Miller, V.A.; et al. Comparative analysis of primary tumour and matched metastases in colorectal cancer patients: Evaluation of concordance between genomic and transcriptional profiles. Eur. J. Cancer 2015, 51, 791–799. [Google Scholar] [CrossRef]
- van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:1802.03426. Available online: https://arxiv.org/pdf/1802.03426 (accessed on 5 July 2021).
- Zhang, Y.; Narayanan, S.P.; Mannan, R.; Raskind, G.; Wang, X.; Vats, P.; Su, F.; Hosseini, N.; Cao, X.; Kumar-Sinha, C.; et al. Single-cell analyses of renal cell cancers reveal insights into tumor microenvironment, cell of origin, and therapy response. Proc. Natl. Acad. Sci. USA 2021, 118, e2103240118. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [Green Version]
- Cillo, A.R.; Kürten, C.H.L.; Tabib, T.; Qi, Z.; Onkar, S.; Wang, T.; Liu, A.; Duvvuri, U.; Kim, S.; Soose, R.J.; et al. Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity 2020, 52, 183–199.e9. [Google Scholar] [CrossRef]
- Zhao, Y.; Pan, Z.; Namburi, S.; Pattison, A.; Posner, A.; Balachander, S.; Paisie, C.A.; Reddi, H.V.; Rueter, J.; Gill, A.J.; et al. CUP-AI-Dx: A tool for inferring cancer tissue of origin and molecular subtype using RNA gene-expression data and artificial intelligence. EBioMedicine 2020, 61, 103030. [Google Scholar] [CrossRef]
- Marquardt, A.; Landwehr, L.-S.; Ronchi, C.L.; Di Dalmazi, G.; Riester, A.; Kollmannsberger, P.; Altieri, B.; Fassnacht, M.; Sbiera, S. Identifying New Potential Biomarkers in Adrenocortical Tumors Based on mRNA Expression Data Using Machine Learning. Cancers 2021, 13, 4671. [Google Scholar] [CrossRef]
- Marquardt, A.; Solimando, A.G.; Kerscher, A.; Bittrich, M.; Kalogirou, C.; Kübler, H.; Rosenwald, A.; Bargou, R.; Kollmannsberger, P.; Schilling, B.; et al. Subgroup-Independent Mapping of Renal Cell Carcinoma-Machine Learning Reveals Prognostic Mitochondrial Gene Signature Beyond Histopathologic Boundaries. Front. Oncol. 2021, 11, 621278. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, H.; Ge, Y.; Wang, X. Integrated single-cell and bulk RNA sequencing analysis identifies a cancer associated fibroblast-related signature for predicting prognosis and therapeutic responses in colorectal cancer. Cancer Cell Int. 2021, 21, 552. [Google Scholar] [CrossRef]
- Kobak, D.; Linderman, G.C. Initialization is critical for preserving global data structure in both t-SNE and UMAP. Nat. Biotechnol. 2021, 39, 156–157. [Google Scholar] [CrossRef]
- Kobak, D.; Linderman, G.; Steinerberger, S.; Kluger, Y.; Berens, P. Heavy-tailed kernels reveal a finer cluster structure in t-SNE visualisations. Mach. Learn. Knowl. Discov. Databases 2020, 11906, 124–139. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagle, N.; Painter, C.; Krevalin, M.; Oh, C.; Anderka, K.; Larkin, K.; Lennon, N.; Dillon, D.; Frank, E.; Winer, E.P.; et al. The Metastatic Breast Cancer Project: A national direct-to-patient initiative to accelerate genomics research. J. Clin. Oncol. 2016, 34, LBA1519. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, H.; Zhang, Y.; Zhang, T.; Gong, J.; Wei, Y.; Duan, Y.-G.; Shu, M.; Yang, Y.; Wu, D.; et al. Dimensionality reduction by UMAP reinforces sample heterogeneity analysis in bulk transcriptomic data. Cell Rep. 2021, 36, 109442. [Google Scholar] [CrossRef] [PubMed]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef]
- Kuksin, M.; Morel, D.; Aglave, M.; Danlos, F.-X.; Marabelle, A.; Zinovyev, A.; Gautheret, D.; Verlingue, L. Applications of single-cell and bulk RNA sequencing in onco-immunology. Eur. J. Cancer 2021, 149, 193–210. [Google Scholar] [CrossRef]
- Traag, V.A.; Waltman, L.; van Eck, N.J. From Louvain to Leiden: Guaranteeing well-connected communities. Sci. Rep. 2019, 9, 5233. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [Green Version]
- Taskesen, E.; Huisman, S.M.H.; Mahfouz, A.; Krijthe, J.H.; de Ridder, J.; van de Stolpe, A.; van den Akker, E.; Verheagh, W.; Reinders, M.J.T. Pan-cancer subtyping in a 2D-map shows substructures that are driven by specific combinations of molecular characteristics. Sci. Rep. 2016, 6, 24949. [Google Scholar] [CrossRef] [Green Version]
- Taskesen, E. Flameplot is a Python Package for the Quantification of Local Similarity across Two Maps or Embeddings. 2020. Available online: https://erdogant.github.io/flameplot (accessed on 18 July 2022).
- Levine, J.H.; Simonds, E.F.; Bendall, S.C.; Davis, K.L.; Amir, E.D.; Tadmor, M.D.; Litvin, O.; Fienberg, H.G.; Jager, A.; Zunder, E.R.; et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef] [Green Version]
- Vidman, L.; Källberg, D.; Rydén, P. Cluster analysis on high dimensional RNA-seq data with applications to cancer research—An evaluation study. PLoS ONE 2019, 14, e0219102. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Butler, W.; Zhou, Y.; Zhang, H.; Tang, L.; Perkinson, K.; Chen, X.; Jiang, X.S.; McCall, S.J.; Inman, B.A.; et al. Pre-existing Castration-resistant Prostate Cancer-like Cells in Primary Prostate Cancer Promote Resistance to Hormonal Therapy. Eur. Urol. 2022, 81, 446–455. [Google Scholar] [CrossRef]
- Pratt, D.; Sahm, F.; Aldape, K. DNA methylation profiling as a model for discovery and precision diagnostics in neuro-oncology. Neuro Oncol. 2021, 23, S16–S29. [Google Scholar] [CrossRef]
- Filipski, K.; Scherer, M.; Zeiner, K.N.; Bucher, A.; Kleemann, J.; Jurmeister, P.; Hartung, T.I.; Meissner, M.; Plate, K.H.; Fenton, T.R.; et al. DNA methylation-based prediction of response to immune checkpoint inhibition in metastatic melanoma. J. Immunother Cancer 2021, 9, e002226. [Google Scholar] [CrossRef]
- Brasó-Maristany, F.; Paré, L.; Chic, N.; Martínez-Sáez, O.; Pascual, T.; Mallafré-Larrosa, M.; Schettini, F.; González-Farré, B.; Sanfeliu, E.; Martínez, D.; et al. Gene expression profiles of breast cancer metastasis according to organ site. Mol. Oncol. 2021, 16, 69–87. [Google Scholar] [CrossRef]
- Cejalvo, J.M.; Martínez de Dueñas, E.; Galván, P.; García-Recio, S.; Burgués Gasión, O.; Paré, L.; Antolín, S.; Martinello, R.; Blancas, I.; Adamo, B.; et al. Intrinsic Subtypes and Gene Expression Profiles in Primary and Metastatic Breast Cancer. Cancer Res. 2017, 77, 2213–2221. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zheng, S.; Truong, L.D.; Ro, J.Y.; Ayala, A.G.; Shen, S.S. Clear cell papillary renal cell carcinoma is the fourth most common histologic type of renal cell carcinoma in 290 consecutive nephrectomies for renal cell carcinoma. Hum. Pathol. 2014, 45, 59–64. [Google Scholar] [CrossRef]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Narayan, A.; Berger, B.; Cho, H. Assessing single-cell transcriptomic variability through density-preserving data visualization. Nat. Biotechnol. 2021, 39, 765–774. [Google Scholar] [CrossRef]
- Lin, W.; Noel, P.; Borazanci, E.H.; Lee, J.; Amini, A.; Han, I.W.; Heo, J.S.; Jameson, G.S.; Fraser, C.; Steinbach, M.; et al. Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions. Genome Med. 2020, 12, 80. [Google Scholar] [CrossRef]
- Pan, H.; Diao, H.; Zhong, W.; Wang, T.; Wen, P.; Wu, C. A Cancer Cell Cluster Marked by LincRNA MEG3 Leads Pancreatic Ductal Adenocarcinoma Metastasis. Front. Oncol. 2021, 11, 656564. [Google Scholar] [CrossRef]
- Xu, K.; Wang, R.; Xie, H.; Hu, L.; Wang, C.; Xu, J.; Zhu, C.; Liu, Y.; Gao, F.; Li, X.; et al. Single-cell RNA sequencing reveals cell heterogeneity and transcriptome profile of breast cancer lymph node metastasis. Oncogenesis 2021, 10, 66. [Google Scholar] [CrossRef]
- Russano, M.; Napolitano, A.; Ribelli, G.; Iuliani, M.; Simonetti, S.; Citarella, F.; Pantano, F.; Dell’Aquila, E.; Anesi, C.; Silvestris, N.; et al. Liquid biopsy and tumor heterogeneity in metastatic solid tumors: The potentiality of blood samples. J. Exp. Clin. Cancer Res. 2020, 39, 95. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marquardt, A.; Kollmannsberger, P.; Krebs, M.; Argentiero, A.; Knott, M.; Solimando, A.G.; Kerscher, A.G. Visual Clustering of Transcriptomic Data from Primary and Metastatic Tumors—Dependencies and Novel Pitfalls. Genes 2022, 13, 1335. https://doi.org/10.3390/genes13081335
Marquardt A, Kollmannsberger P, Krebs M, Argentiero A, Knott M, Solimando AG, Kerscher AG. Visual Clustering of Transcriptomic Data from Primary and Metastatic Tumors—Dependencies and Novel Pitfalls. Genes. 2022; 13(8):1335. https://doi.org/10.3390/genes13081335
Chicago/Turabian StyleMarquardt, André, Philip Kollmannsberger, Markus Krebs, Antonella Argentiero, Markus Knott, Antonio Giovanni Solimando, and Alexander Georg Kerscher. 2022. "Visual Clustering of Transcriptomic Data from Primary and Metastatic Tumors—Dependencies and Novel Pitfalls" Genes 13, no. 8: 1335. https://doi.org/10.3390/genes13081335
APA StyleMarquardt, A., Kollmannsberger, P., Krebs, M., Argentiero, A., Knott, M., Solimando, A. G., & Kerscher, A. G. (2022). Visual Clustering of Transcriptomic Data from Primary and Metastatic Tumors—Dependencies and Novel Pitfalls. Genes, 13(8), 1335. https://doi.org/10.3390/genes13081335