
A Primer Genetic Toolkit for Exploring Mitochondrial Biology and Disease Using Zebrafish

 , ,
, ,  ,
, 

Abstract
1. Introduction
2. Methods
2.1. Zebrafish Handling
2.2. Identification of Zebrafish Orthologs Having a Putative Mitochondrial Function
2.3. Mutant Generation
2.4. TALEN Design/Assembly/Delivery
2.5. TALEN Mutant Screening
2.6. Gene Breaking Transposon System
2.7. In Silico Analysis to Determine Protein Homology and Alteration of DNA Sequence by TALENs and the GBT in Zebrafish Mutants
2.8. RNA Extraction and Sample Preparation
2.9. RNA Sequencing and Analysis
2.10. Access to All Reported Reagents—Zebrafish and Sequences
3. Results
3.1. Generation of Zebrafish Mutant Collection
3.2. TALEN- and GBT-Mediated Targeting of Nuclear-Encoded Mitochondrial Genes
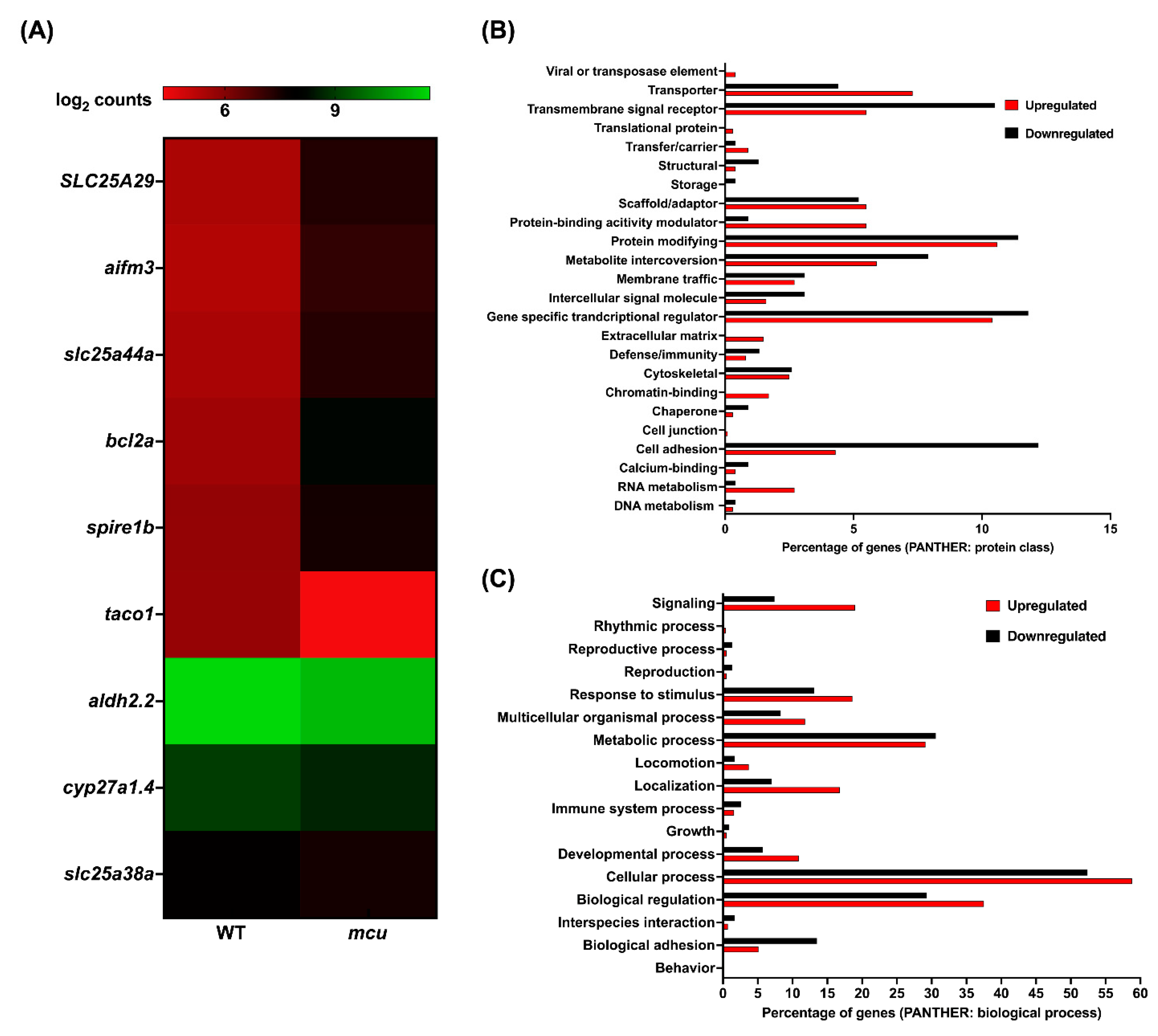
3.3. Mitochondrial Zebrafish Mutants Display Altered Transcriptomic Signature
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Martin, W. The genome of Rickettsia prowazekii and some thoughts on the origin of mitochondria and hydrogenosomes. Bioessays 1999, 21, 377–381. [Google Scholar] [CrossRef]
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Koskella, B.; Schaack, S. Mutation pressure and the evolution of organelle genomic architecture. Science 2006, 311, 1727–1730. [Google Scholar] [CrossRef]
- Thorsness, P.E.; Weber, E.R. Escape and migration of nucleic acids between chloroplasts, mitochondria, and the nucleus. Int. Rev. Cytol. 1996, 165, 207–234. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 2010, 51, 440–450. [Google Scholar] [CrossRef]
- Goldstein, A.; Falk, M.J. Mitochondrial DNA Deletion Syndromes. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; 1993. [Google Scholar]
- Elson, J.L.; Smith, P.M.; Greaves, L.C.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.; Taylor, R.W.; Vila-Sanjurjo, A. The presence of highly disruptive 16S rRNA mutations in clinical samples indicates a wider role for mutations of the mitochondrial ribosome in human disease. Mitochondrion 2015, 25, 17–27. [Google Scholar] [CrossRef]
- Elson, J.L.; Samuels, D.C.; Turnbull, D.M.; Chinnery, P.F. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am. J. Hum. Genet. 2001, 68, 802–806. [Google Scholar] [CrossRef]
- Broughton, R.E.; Milam, J.E.; Roe, B.A. The complete sequence of the zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA. Genome Res. 2001, 11, 1958–1967. [Google Scholar] [CrossRef]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef] [PubMed]
- Richter, R.; Pajak, A.; Dennerlein, S.; Rozanska, A.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Translation termination in human mitochondrial ribosomes. Biochem. Soc. Trans. 2010, 38, 1523–1526. [Google Scholar] [CrossRef] [PubMed]
- Osawa, S.; Jukes, T.H.; Watanabe, K.; Muto, A. Recent evidence for evolution of the genetic code. Microbiol. Rev. 1992, 56, 229–264. [Google Scholar] [CrossRef]
- Srivastava, S.; Moraes, C.T. Double-strand breaks of mouse muscle mtDNA promote large deletions similar to multiple mtDNA deletions in humans. Hum. Mol. Genet. 2005, 14, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Minczuk, M.; Moraes, C.T. Mechanisms of Mitochondrial DNA Deletion Formation. Trends Genet. 2019, 35, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Bayona-Bafaluy, M.P.; Blits, B.; Battersby, B.J.; Shoubridge, E.A.; Moraes, C.T. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc. Natl. Acad. Sci. USA 2005, 102, 14392–14397. [Google Scholar] [CrossRef] [PubMed]
- Zinovkina, L.A. Mechanisms of Mitochondrial DNA Repair in Mammals. Biochemistry 2018, 83, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—Critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef]
- Patananan, A.N.; Wu, T.H.; Chiou, P.Y.; Teitell, M.A. Modifying the Mitochondrial Genome. Cell Metab. 2016, 23, 785–796. [Google Scholar] [CrossRef]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef]
- Mok, B.Y.; Kotrys, A.V.; Raguram, A.; Huang, T.P.; Mootha, V.K.; Liu, D.R. CRISPR-free base editors with enhanced activity and expanded targeting scope in mitochondrial and nuclear DNA. Nat. Biotechnol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhang, X.; Chen, X.; Sun, H.; Dai, Y.; Wang, J.; Qian, X.; Tan, L.; Lou, X.; Shen, B. Precision modeling of mitochondrial diseases in zebrafish via DdCBE-mediated mtDNA base editing. Cell Discov. 2021, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, A.; Kar, B.; Restrepo-Castillo, S.; Holmberg, S.R.; Mathew, N.D.; Kendall, B.L.; Cotter, R.P.; WareJoncas, Z.; Seiler, C.; Nakamaru-Ogiso, E.; et al. The FusX TALE Base Editor (FusXTBE) for Rapid Mitochondrial DNA Programming of Human Cells In Vitro and Zebrafish Disease Models In Vivo. CRISPR J. 2021, 4, 799–821. [Google Scholar] [CrossRef]
- Cho, S.I.; Lee, S.; Mok, Y.G.; Lim, K.; Lee, J.; Lee, J.M.; Chung, E.; Kim, J.S. Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell 2022, 185, 1764–1776. [Google Scholar] [CrossRef]
- Lee, H.; Lee, S.; Baek, G.; Kim, A.; Kang, B.C.; Seo, H.; Kim, J.S. Mitochondrial DNA editing in mice with DddA-TALE fusion deaminases. Nat. Commun. 2021, 12, 1190. [Google Scholar] [CrossRef]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Lopez, M.F.; Kristal, B.S.; Chernokalskaya, E.; Lazarev, A.; Shestopalov, A.I.; Bogdanova, A.; Robinson, M. High-throughput profiling of the mitochondrial proteome using affinity fractionation and automation. Electrophoresis 2000, 21, 3427–3440. [Google Scholar] [CrossRef]
- Elstner, M.; Andreoli, C.; Klopstock, T.; Meitinger, T.; Prokisch, H. The mitochondrial proteome database: MitoP2. Methods Enzymol. 2009, 457, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent Advances in Mitochondrial Disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska, M.; Gorman, G.S.; Alston, C.L.; Pajak, A.; Pyle, A.; He, L.; Griffin, H.; Chinnery, P.F.; Miller, J.A.; Schaefer, A.M.; et al. Adult Onset Leigh Syndrome in the Intensive Care Setting: A Novel Presentation of a C12orf65 Related Mitochondrial Disease. J. Neuromuscul. Dis. 2015, 2, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Shehata, B.M.; Cundiff, C.A.; Lee, K.; Sabharwal, A.; Lalwani, M.K.; Davis, A.K.; Agrawal, V.; Sivasubbu, S.; Iannucci, G.J.; Gibson, G. Exome sequencing of patients with histiocytoid cardiomyopathy reveals a de novo NDUFB11 mutation that plays a role in the pathogenesis of histiocytoid cardiomyopathy. Am. J. Med. Genet. A 2015, 167, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef]
- Ghezzi, D.; Saada, A.; D’Adamo, P.; Fernandez-Vizarra, E.; Gasparini, P.; Tiranti, V.; Elpeleg, O.; Zeviani, M. FASTKD2 nonsense mutation in an infantile mitochondrial encephalomyopathy associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 2008, 83, 415–423. [Google Scholar] [CrossRef]
- Floyd, B.J.; Wilkerson, E.M.; Veling, M.T.; Minogue, C.E.; Xia, C.; Beebe, E.T.; Wrobel, R.L.; Cho, H.; Kremer, L.S.; Alston, C.L.; et al. Mitochondrial Protein Interaction Mapping Identifies Regulators of Respiratory Chain Function. Mol. Cell 2016, 63, 621–632. [Google Scholar] [CrossRef]
- Altmann, K.; Durr, M.; Westermann, B. Saccharomyces cerevisiae as a model organism to study mitochondrial biology: General considerations and basic procedures. Methods Mol. Biol. 2007, 372, 81–90. [Google Scholar] [CrossRef]
- Denoth Lippuner, A.; Julou, T.; Barral, Y. Budding yeast as a model organism to study the effects of age. FEMS Microbiol. Rev. 2014, 38, 300–325. [Google Scholar] [CrossRef]
- Schwimmer, C.; Rak, M.; Lefebvre-Legendre, L.; Duvezin-Caubet, S.; Plane, G.; di Rago, J.P. Yeast models of human mitochondrial diseases: From molecular mechanisms to drug screening. Biotechnol. J. 2006, 1, 270–281. [Google Scholar] [CrossRef]
- Addo, M.G.; Cossard, R.; Pichard, D.; Obiri-Danso, K.; Rotig, A.; Delahodde, A. Caenorhabditis elegans, a pluricellular model organism to screen new genes involved in mitochondrial genome maintenance. Biochim. Biophys. Acta 2010, 1802, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Arriola, E.; Cardenas-Rodriguez, N.; Coballase-Urrutia, E.; Pedraza-Chaverri, J.; Carmona-Aparicio, L.; Ortega-Cuellar, D. Caenorhabditis elegans: A useful model for studying metabolic disorders in which oxidative stress is a contributing factor. Oxid. Med. Cell. Longev. 2014, 2014, 705253. [Google Scholar] [CrossRef] [PubMed]
- Tissenbaum, H.A. Using C. elegans for aging research. Invertebr. Reprod. Dev. 2015, 59, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Moreno, M.A.; Farr, C.L.; Kaguni, L.S.; Garesse, R. Drosophila melanogaster as a model system to study mitochondrial biology. Methods Mol. Biol. 2007, 372, 33–49. [Google Scholar] [CrossRef]
- Guo, M. Drosophila as a model to study mitochondrial dysfunction in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009944. [Google Scholar] [CrossRef]
- Sanchez-Martinez, A.; Luo, N.; Clemente, P.; Adan, C.; Hernandez-Sierra, R.; Ochoa, P.; Fernandez-Moreno, M.A.; Kaguni, L.S.; Garesse, R. Modeling human mitochondrial diseases in flies. Biochim. Biophys. Acta 2006, 1757, 1190–1198. [Google Scholar] [CrossRef]
- Sen, A.; Cox, R.T. Fly Models of Human Diseases: Drosophila as a Model for Understanding Human Mitochondrial Mutations and Disease. Curr. Top. Dev. Biol. 2017, 121, 1–27. [Google Scholar] [CrossRef]
- Boumans, H.; Grivell, L.A.; Berden, J.A. The respiratory chain in yeast behaves as a single functional unit. J. Biol. Chem. 1998, 273, 4872–4877. [Google Scholar] [CrossRef]
- Brown, W.M.; George, M., Jr.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef]
- Gershoni, M.; Fuchs, A.; Shani, N.; Fridman, Y.; Corral-Debrinski, M.; Aharoni, A.; Frishman, D.; Mishmar, D. Coevolution predicts direct interactions between mtDNA-encoded and nDNA-encoded subunits of oxidative phosphorylation complex i. J. Mol. Biol. 2010, 404, 158–171. [Google Scholar] [CrossRef]
- Grossman, L.I.; Wildman, D.E.; Schmidt, T.R.; Goodman, M. Accelerated evolution of the electron transport chain in anthropoid primates. Trends Genet. 2004, 20, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Haen, K.M.; Pett, W.; Lavrov, D.V. Parallel loss of nuclear-encoded mitochondrial aminoacyl-tRNA synthetases and mtDNA-encoded tRNAs in Cnidaria. Mol. Biol. Evol. 2010, 27, 2216–2219. [Google Scholar] [CrossRef] [PubMed]
- Kuzmenko, A.V.; Levitskii, S.A.; Vinogradova, E.N.; Atkinson, G.C.; Hauryliuk, V.; Zenkin, N.; Kamenski, P.A. Protein biosynthesis in mitochondria. Biochemistry 2013, 78, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V.; Pett, W. Animal Mitochondrial DNA as We Do Not Know It: Mt-Genome Organization and Evolution in Nonbilaterian Lineages. Genome Biol. Evol. 2016, 8, 2896–2913. [Google Scholar] [CrossRef]
- Lightowlers, R.N.; Rozanska, A.; Chrzanowska-Lightowlers, Z.M. Mitochondrial protein synthesis: Figuring the fundamentals, complexities and complications, of mammalian mitochondrial translation. FEBS Lett. 2014, 588, 2496–2503. [Google Scholar] [CrossRef]
- Pett, W.; Lavrov, D.V. Cytonuclear Interactions in the Evolution of Animal Mitochondrial tRNA Metabolism. Genome Biol. Evol. 2015, 7, 2089–2101. [Google Scholar] [CrossRef]
- Rea, S.L.; Graham, B.H.; Nakamaru-Ogiso, E.; Kar, A.; Falk, M.J. Bacteria, yeast, worms, and flies: Exploiting simple model organisms to investigate human mitochondrial diseases. Dev. Disabil. Res. Rev. 2010, 16, 200–218. [Google Scholar] [CrossRef]
- Zhao, J.; Lendahl, U.; Nister, M. Regulation of mitochondrial dynamics: Convergences and divergences between yeast and vertebrates. Cell Mol. Life Sci. 2013, 70, 951–976. [Google Scholar] [CrossRef]
- Blanco-Sanchez, B.; Clement, A.; Phillips, J.B.; Westerfield, M. Zebrafish models of human eye and inner ear diseases. Methods Cell Biol. 2017, 138, 415–467. [Google Scholar] [CrossRef]
- Lin, C.Y.; Chiang, C.Y.; Tsai, H.J. Zebrafish and Medaka: New model organisms for modern biomedical research. J. Biomed. Sci. 2016, 23, 19. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Fichi, G.; Naef, V.; Barca, A.; Longo, G.; Fronte, B.; Verri, T.; Santorelli, F.M.; Marchese, M.; Petruzzella, V. Fishing in the Cell Powerhouse: Zebrafish as A Tool for Exploration of Mitochondrial Defects Affecting the Nervous System. Int. J. Mol. Sci. 2019, 20, 2409. [Google Scholar] [CrossRef]
- Steele, S.L.; Prykhozhij, S.V.; Berman, J.N. Zebrafish as a model system for mitochondrial biology and diseases. Transl. Res. 2014, 163, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kang, K.H.; Kim, C.H.; Choi, S.Y. Real-time imaging of mitochondria in transgenic zebrafish expressing mitochondrially targeted GFP. Biotechniques 2008, 45, 331–334. [Google Scholar] [CrossRef]
- Pinho, B.R.; Santos, M.M.; Fonseca-Silva, A.; Valentao, P.; Andrade, P.B.; Oliveira, J.M. How mitochondrial dysfunction affects zebrafish development and cardiovascular function: An in vivo model for testing mitochondria-targeted drugs. Br. J. Pharmacol. 2013, 169, 1072–1090. [Google Scholar] [CrossRef] [PubMed]
- Polyak, E.; Ostrovsky, J.; Peng, M.; Dingley, S.D.; Tsukikawa, M.; Kwon, Y.J.; McCormack, S.E.; Bennett, M.; Xiao, R.; Seiler, C.; et al. N-acetylcysteine and vitamin E rescue animal longevity and cellular oxidative stress in pre-clinical models of mitochondrial complex I disease. Mol. Genet. Metab. 2018, 123, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef]
- Byrnes, J.; Ganetzky, R.; Lightfoot, R.; Tzeng, M.; Nakamaru-Ogiso, E.; Seiler, C.; Falk, M.J. Pharmacologic modeling of primary mitochondrial respiratory chain dysfunction in zebrafish. Neurochem. Int. 2018, 117, 23–34. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, M.; Hu, B.; Huang, R.; Hu, B. In vivo Imaging of Mitochondrial Transport in Single-Axon Regeneration of Zebrafish Mauthner Cells. Front. Cell. Neurosci. 2017, 11, 4. [Google Scholar] [CrossRef]
- Dowling, J.J.; Arbogast, S.; Hur, J.; Nelson, D.D.; McEvoy, A.; Waugh, T.; Marty, I.; Lunardi, J.; Brooks, S.V.; Kuwada, J.Y.; et al. Oxidative stress and successful antioxidant treatment in models of RYR1-related myopathy. Brain 2012, 135, 1115–1127. [Google Scholar] [CrossRef]
- Song, Y.; Selak, M.A.; Watson, C.T.; Coutts, C.; Scherer, P.C.; Panzer, J.A.; Gibbs, S.; Scott, M.O.; Willer, G.; Gregg, R.G.; et al. Mechanisms underlying metabolic and neural defects in zebrafish and human multiple acyl-CoA dehydrogenase deficiency (MADD). PLoS One 2009, 4, e8329. [Google Scholar] [CrossRef] [PubMed]
- Pickett, S.B.; Thomas, E.D.; Sebe, J.Y.; Linbo, T.; Esterberg, R.; Hailey, D.W.; Raible, D.W. Cumulative mitochondrial activity correlates with ototoxin susceptibility in zebrafish mechanosensory hair cells. Elife 2018, 7, e38062. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Pinter, K.; Drerup, C.M. Analyzing Neuronal Mitochondria in vivo Using Fluorescent Reporters in Zebrafish. Front. Cell Dev. Biol. 2018, 6, 144. [Google Scholar] [CrossRef] [PubMed]
- Plucinska, G.; Paquet, D.; Hruscha, A.; Godinho, L.; Haass, C.; Schmid, B.; Misgeld, T. In vivo imaging of disease-related mitochondrial dynamics in a vertebrate model system. J. Neurosci. 2012, 32, 16203–16212. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.L.; Upadhyayula, S.; Milkie, D.E.; Singh, V.; Wang, K.; Swinburne, I.A.; Mosaliganti, K.R.; Collins, Z.M.; Hiscock, T.W.; Shea, J.; et al. Observing the cell in its native state: Imaging subcellular dynamics in multicellular organisms. Science 2018, 360. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.C.; McNulty, M.S.; Poshusta, T.L.; Campbell, J.M.; Martinez-Galvez, G.; Argue, D.P.; Lee, H.B.; Urban, M.D.; Bullard, C.E.; Blackburn, P.R.; et al. FusX: A Rapid One-Step Transcription Activator-Like Effector Assembly System for Genome Science. Hum. Gene Ther. 2016, 27, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.J.; Balciunas, D.; Pogoda, H.M.; Ding, Y.; Westcot, S.E.; Bedell, V.M.; Greenwood, T.M.; Urban, M.D.; Skuster, K.J.; Petzold, A.M.; et al. In vivo protein trapping produces a functional expression codex of the vertebrate proteome. Nat. Methods 2011, 8, 506–515. [Google Scholar] [CrossRef]
- Ichino, N.; Serres, M.R.; Urban, R.M.; Urban, M.D.; Treichel, A.J.; Schaefbauer, K.J.; Greif, L.E.; Varshney, G.K.; Skuster, K.J.; McNulty, M.S.; et al. Building the vertebrate codex using the gene breaking protein trap library. Elife 2020, 9, e54572. [Google Scholar] [CrossRef]
- Neff, K.L.; Argue, D.P.; Ma, A.C.; Lee, H.B.; Clark, K.J.; Ekker, S.C. Mojo Hand, a TALEN design tool for genome editing applications. BMC Bioinform. 2013, 14, 1. [Google Scholar] [CrossRef]
- Kendziorski, C.; Irizarry, R.A.; Chen, K.S.; Haag, J.D.; Gould, M.N. On the utility of pooling biological samples in microarray experiments. Proc. Natl. Acad. Sci. USA 2005, 102, 4252–4257. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Gerstner, N.; Kehl, T.; Lenhof, K.; Muller, A.; Mayer, C.; Eckhart, L.; Grammes, N.L.; Diener, C.; Hart, M.; Hahn, O.; et al. GeneTrail 3: Advanced high-throughput enrichment analysis. Nucleic Acids Res. 2020, 48, W515–W520. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Clark, K.J.; Argue, D.P.; Petzold, A.M.; Ekker, S.C. zfishbook: Connecting you to a world of zebrafish revertible mutants. Nucleic Acids Res. 2012, 40, D907–D911. [Google Scholar] [CrossRef][Green Version]
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial respiratory complex I: Structure, function and implication in human diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef]
- King, M.P.; Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 1989, 246, 500–503. [Google Scholar] [CrossRef]
- Koga, Y.; Davidson, M.; Schon, E.A.; King, M.P. Analysis of cybrids harboring MELAS mutations in the mitochondrial tRNA(Leu(UUR)) gene. Muscle Nerve Suppl. 1995, 3, S119–S123. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Carl, S.M.; Swerdlow, R.H. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2014, 2, 619–631. [Google Scholar] [CrossRef]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Bedell, V.M.; Wang, Y.; Campbell, J.M.; Poshusta, T.L.; Starker, C.G.; Krug, R.G., 2nd; Tan, W.; Penheiter, S.G.; Ma, A.C.; Leung, A.Y.; et al. In vivo genome editing using a high-efficiency TALEN system. Nature 2012, 491, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, A.; Wishman, M.D.; Cervera, R.L.; Serres, M.R.; Anderson, J.L.; Treichel, A.J.; Ichino, N.; Liu, W.; Yang, J.; Ding, Y.; et al. A Genetic Model Therapy Proposes a Critical Role for Liver Dysfunction in Mitochondrial Biology and Disease. bioRxiv 2000, 2020.2005.2008.084681. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approved Human Symbol | Approved Name | HUMAN chr | Zebrafish Orthologs | Zebrafish chr | Allele Designator | Mouse Orthologs | Mouse chr | Clinical Phenotype Observed in Humans | Disease | Biological Function | OMIM ID |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutants created by Transcription activator-like effector nucleases (TALEN) | |||||||||||
| NDUFAF6 | NADH:ubiquinone oxidoreductase complex assembly factor 6 | 8 | ndufaf6 | 16 | mn0117 | Ndufaf6 | 4 | Focal right-hand seizures, ataxia, lactic acidosis | Leigh syndrome, mitochondrial com-plex I deficiency, nuclear type 17 | Oxidative phosphorylation complex assembly—Complex I | 612392 |
| NDUFS4 | NADH: ubiquinone oxidoreductase subunit S4 | 5 | ndufs4 | 5 | mn0118 | Ndufs4 | 13 | Mitochondrial complex-I deficiency | Mitochondrial complex-I deficiency, nuclear type 1 | Oxidative phosphorylation enzymes—Complex I | 602694 |
| SDHA | Succinate dehydrogenase complex flavoprotein subunit A | 5 | sdha | 19 | mn0121 | Sdha | 13 | Cardiomegaly, cardiomyopathy, nys-tagmus, hypotonia, gastrointestinal stromal tumors, paragangliomas, pheochromocytoma, psychomotor regression | Mitochondrial complex-II deficien-cy, nuclear type 1; Cardiomyopathy dilated, 1GG, Leigh syndrome; Paragan-glioma | Oxidative phosphorylation enzymes—Complex II | 600857 |
| COQ2 | Coenzyme Q2, polyprenyltransferase | 4 | coq2 | 5 | mn0106 | Coq2 | 5 | Multiple system atrophy, unsteadiness of gait, nystagmus, gait ataxia, dysarthria, speech difficulty, dysmetria, lactic acidosis, renal dysfunction, and nystagmus | Coenzyme Q10 deficiency, primary; Multiple system atrophy | Biosynthesis of CoQ, coenzyme in mitochondrial respiratory chain | 609825 |
| PDSS2 | Decaprenyl-diphosphate synthase subunit 2 | 6 | pdss2 | 13 | mn0120 | Pdss2 | 10 | Hypotonia, seizures, cortical blindness, lactic acidosis, encephalopathy, and nephrotic syndrome | Coenzyme Q10 defi-ciency, primary 3 | Involved in biosynthesis of coenzyme Q | 610564 |
| UQCRQ | Ubiquinol- cytochrome c reductase complex III subunit VII | 5 | uqcrq | 14 | mn0128 | Uqcrq | 11 | Severe psychomotor retardation and extrapyramidal signs, dystonia, athetosis, ataxia, mild axial hypotonia and marked global dementia | Mitochondrial com-plex III deficiency, nuclear type 4 | Mitochondrial complex III: ubiquinol—cytochrome c reductase complex subunits | 612080 |
| SURF1 | SURF1 cytochrome c oxidase assembly factor | 9 | surf1 | 5 | mn0123 | Surf1 | 2 | Childhood onset neuropathy, lactic acidosis, mild intellectual disability ataxia, facial dysmorphism, encephalopathy, hypotonia, cerebellar ataxia, deafness, ophthalmoplegia, growth retardation and nystagmus | Charcot-Marie- Tooth disease, type 4K; Leigh syndrome, due to COX IV deficiency | Involved in biogenesis of cytochrome c oxidase complex | 185620 |
| COX10 | Cytochrome c oxidase assembly factor heme A: farnesyltransferase COX10 | 17 | cox10 | 12 | mn0107 | Cox10 | 11 | Muscle weakness, hypotonia, ataxia, ptosis, pyramidal syndrome, status epilepticus, lactic acidosis, hypertrophic cardiomyopathy, hypoglycemia | Leigh syndrome due to mitochondrial COX IV, nuclear type 3 deficiency; mito-chondrial COX IV deficiency | Oxidative phosphorylation complex assembly—Complex IV | 602125 |
| TMEM70 | Transmembrane protein 70 | 8 | tmem70 | 2 | mn0126 mn0127 | Tmem70 | 1 | Lactic acidosis, encephalopathy, mi-crocephaly, hypotonia, facial dys-morphism and 3- methylglutaconic aciduria psychomotor delay and hy-perammonemia | Mitochondrial complex V (ATP synthase) deficiency, nuclear type 2 | Biogenesis of mitochondrial ATP synthase | 612418 |
| ATP5F1E | ATP synthase F1 subunit epsilon | 20 | atp5f1e | 6 | mn0101 | Atp5f1e | 2 | Neonatal-onset lactic acidosis, 3- methylglutaconic aciduria, mild mental retardation, and peripheral neuropathy | Mitochondrial com-plex (ATP synthase) deficiency, nuclear type 3 | Oxidative phosphorylation complex assembly—ATP synthase | 606153 |
| MCU | Mitochondrial calcium uniporter | 10 | mcu | 13 | mn0111 | Mcu | 10 | n/a | n/a | Calcium transporter and mediates calcium uptake in mitochondria | 614197 |
| MICU1 | Mitochondrial calcium uptake 1 | 10 | micu1 | 13 | mn0132 | Micu1 | 10 | Proximal muscle weakness and learning disabilities | Myopathy with extrapyramidal signs | Regulator of mitochondrial calcium uptake | 605084 |
| MTFMT | Mitochondrial methionyl-tRNA formyltransferase | 15 | mtfmt | 7 | mn0114 mn0115 | Mtfmt | 9 | Psychomotor developmental delay, renal dysplasia, dysarthria and ataxia | Combined oxidative phosphorylation deficiency | Catalyzes the formylation of methionyl—tRNA | 611766 |
| Mutants created by Gene-breaking transposon (GBT) | |||||||||||
| MCU | Mitochondrial calcium uniporter | 10 | mcu | 13 | mn0599 | Mcu | 10 | n/a | n/a | Calcium transporter and mediates calcium uptake in mitochondria | 614197 |
| LRPPRC | Leucine rich pentatricopeptide repeat containing | 2 | lrpprc | 13 | mn0235gt | Lrpprc | 17 | Delayed psychomotor development, mental retardation, mild dysmorphic facial features, hypotonia, ataxia, sei-zures, dysphagia, and hypertrophic cardiomyopathy | Leigh syndrome, French-Canadian type | Involved in translation of mitochondrial encoded cox subunits and mediation of folding of mitochondrial transcriptome | 607544 |
| MRPS18B | Mitochondrial ribosomal protein S18B | 6 | mrps18b | 19 | mn0425gt | Mrps18b | 17 | n/a | n/a | Part of small 28S subunit of mitoribosome | 611982 |
| TIMM50 | Translocase of inner mitochondrial membrane 50 | 19 | timm50 | 15 | mn0906gt | Timm50 | 7 | Severe intellectual disability, seizure and 3- methylglutaconic aciduria | 3-methylglutaconic aciduria, type IX; Mitochondrial complex V deficiency | Subunit of TIM23 inner mitochondrial membrane complex and recognizes mitochondrial targeting signal or pre-sequence | 607381 |
| OGDH | Oxoglutarate dehydrogenase | 7 | ogdhb | 10 | mn0281gt | Ogdh | 11 | Colorectal cancer | Alpha- ketoglutarate dehydrogenase deficiency | Catalyzes the conversion of 2-oxoglutarate to succinyl-CoA and CO2 | 613022 |
| IDH2 | Isocitrate dehydrogenase (NADP (+)) 2, mitochondrial | 15 | idh2 | 18 | mn1651gt | Idh2 | 7 | Acute myeloid leukemia and abnormal production of D-2-hydroxyglutaric acid | D-2- hydroxyglutaric aciduria 2 | Catalyzes the conversion of isocitrate to 2- oxoglutarate | 147650 |
| CASQ1 | Calsequestrin | 1 | casq1a | 2 | mn0437gt | Casq1 | 1 | Vacuolar myopathy | Myopathy, vacuolar, with CASQ1 aggregates | Luminal sarcoplasmic reticulum calcium sensor | 114250 |
| DELE1 | DAP3 binding cell death enhancer 1 | 5 | dele1 | 14 | mn0926gt | Dele1 | 18 | n/a | n/a | Key activator of the integrated stress response (ISR) following mitochondrial stress | 615741 |
| NSUN2* | NOP2/Sun RNA methyltransferase 2 | 5 | nsun2 | 19 | mn1104gt* | Nsun2 | 13 | Intellectual developmental disorder-5, some mild dysmorphic features, including microcephaly, long and narrow face, bushy eyebrows with synophrys, hypotelorism, large nose with long columella and hypoplastic alae nasi, short philtrum, and full upper lip, later onset of muscular hypertonia and spasticity | Intellectual developmental disorder, autosomal recessive 5 | Methyltransferase that catalyzes the methylation of cytosine to 5-methylcytosine (m5C) at position 34 of intron-containing tRNA (Leu)(CAA) precursor | 610916 |
| LAP3* | Leucine aminopeptidase 3 | 4 | lap3 | 1 | mn0582gt* | Lap3 | 5 | n/a | n/a | Enable peptidase activity | 170250 |
| D2HGDH | D-2-hydroxyglutarate dehydrogenase | 2 | d2hgdh | 2 | mn1118gt | D2hgdh | 1 | Tonic-clonic, and myoclonic seizures, D-2-hydroxyglutaric aciduria | D-2-hydroxyglutaric aciduria | Enables D-2hydroxyglutarate dehydrogenase, belonging to the FAD-binding oxidoreductase/transferase type 4 family | 609186 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabharwal, A.; Campbell, J.M.; Schwab, T.L.; WareJoncas, Z.; Wishman, M.D.; Ata, H.; Liu, W.; Ichino, N.; Hunter, D.E.; Bergren, J.D.; et al. A Primer Genetic Toolkit for Exploring Mitochondrial Biology and Disease Using Zebrafish. Genes 2022, 13, 1317. https://doi.org/10.3390/genes13081317
Sabharwal A, Campbell JM, Schwab TL, WareJoncas Z, Wishman MD, Ata H, Liu W, Ichino N, Hunter DE, Bergren JD, et al. A Primer Genetic Toolkit for Exploring Mitochondrial Biology and Disease Using Zebrafish. Genes. 2022; 13(8):1317. https://doi.org/10.3390/genes13081317
Chicago/Turabian StyleSabharwal, Ankit, Jarryd M. Campbell, Tanya L. Schwab, Zachary WareJoncas, Mark D. Wishman, Hirotaka Ata, Wiebin Liu, Noriko Ichino, Danielle E. Hunter, Jake D. Bergren, and et al. 2022. "A Primer Genetic Toolkit for Exploring Mitochondrial Biology and Disease Using Zebrafish" Genes 13, no. 8: 1317. https://doi.org/10.3390/genes13081317
APA StyleSabharwal, A., Campbell, J. M., Schwab, T. L., WareJoncas, Z., Wishman, M. D., Ata, H., Liu, W., Ichino, N., Hunter, D. E., Bergren, J. D., Urban, M. D., Urban, R. M., Holmberg, S. R., Kar, B., Cook, A., Ding, Y., Xu, X., Clark, K. J., & Ekker, S. C. (2022). A Primer Genetic Toolkit for Exploring Mitochondrial Biology and Disease Using Zebrafish. Genes, 13(8), 1317. https://doi.org/10.3390/genes13081317