
Genome-Wide Association Study Identified a Quantitative Trait Locus and Two Candidate Genes on Sus scrofa Chromosome 2 Affecting Vulvar Traits of Suhuai Pigs

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Animals and Phenotypic Collection
2.3. Genotyping and Imputation
2.4. Descriptive Analyses and Heritability Estimation
2.5. Genome-Wide Association Study
2.6. Linkage Disequilibrium and Linkage Analysis (LDLA)
2.7. Phenotypic Variation Explained by SNPs
2.8. Annotation of Candidate Genes
3. Results
3.1. Descriptive Results of VL, VW and VA in Suhuai Pigs
3.2. Imputation Description
3.3. The Test of Population Stratification
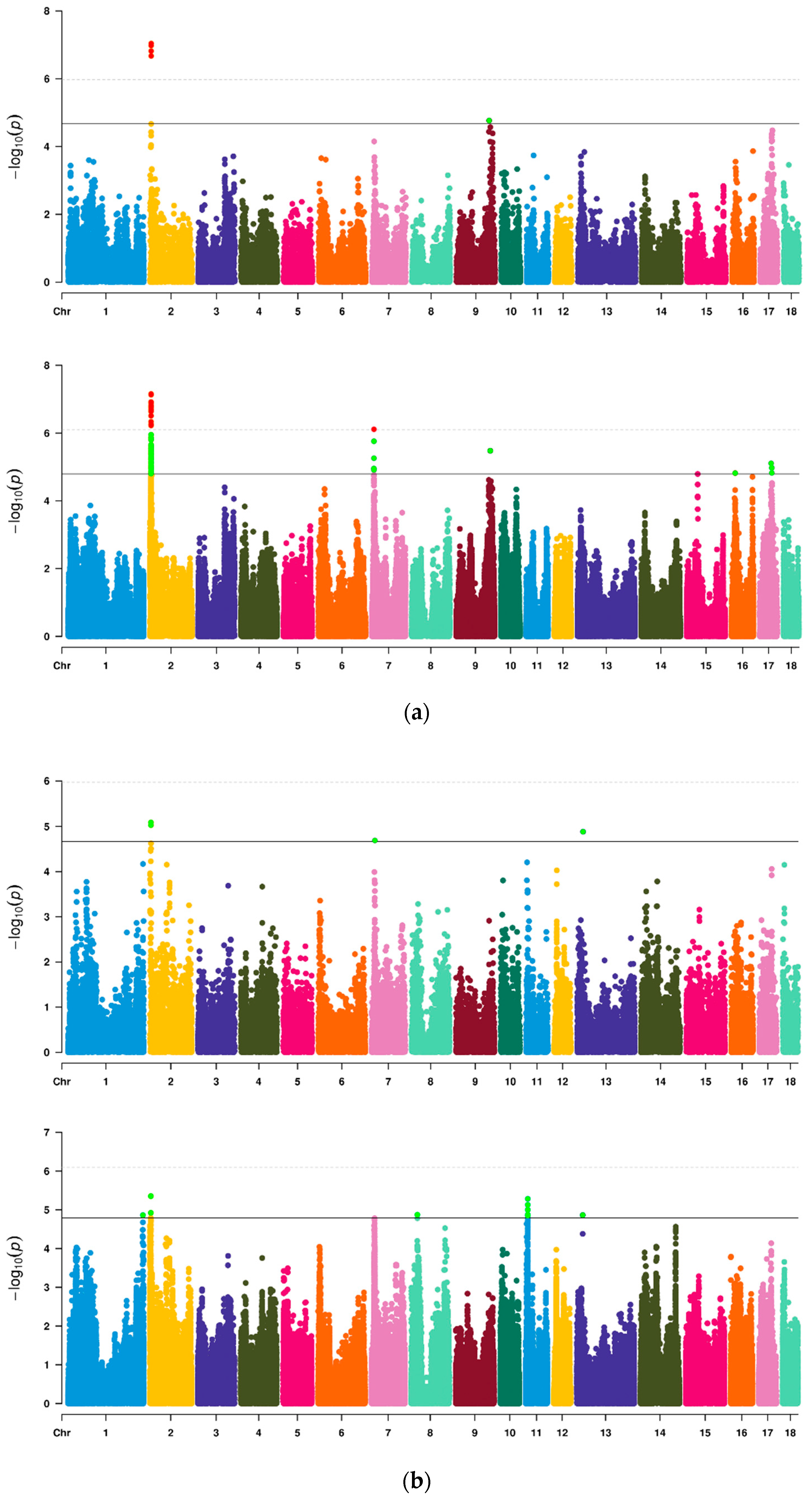
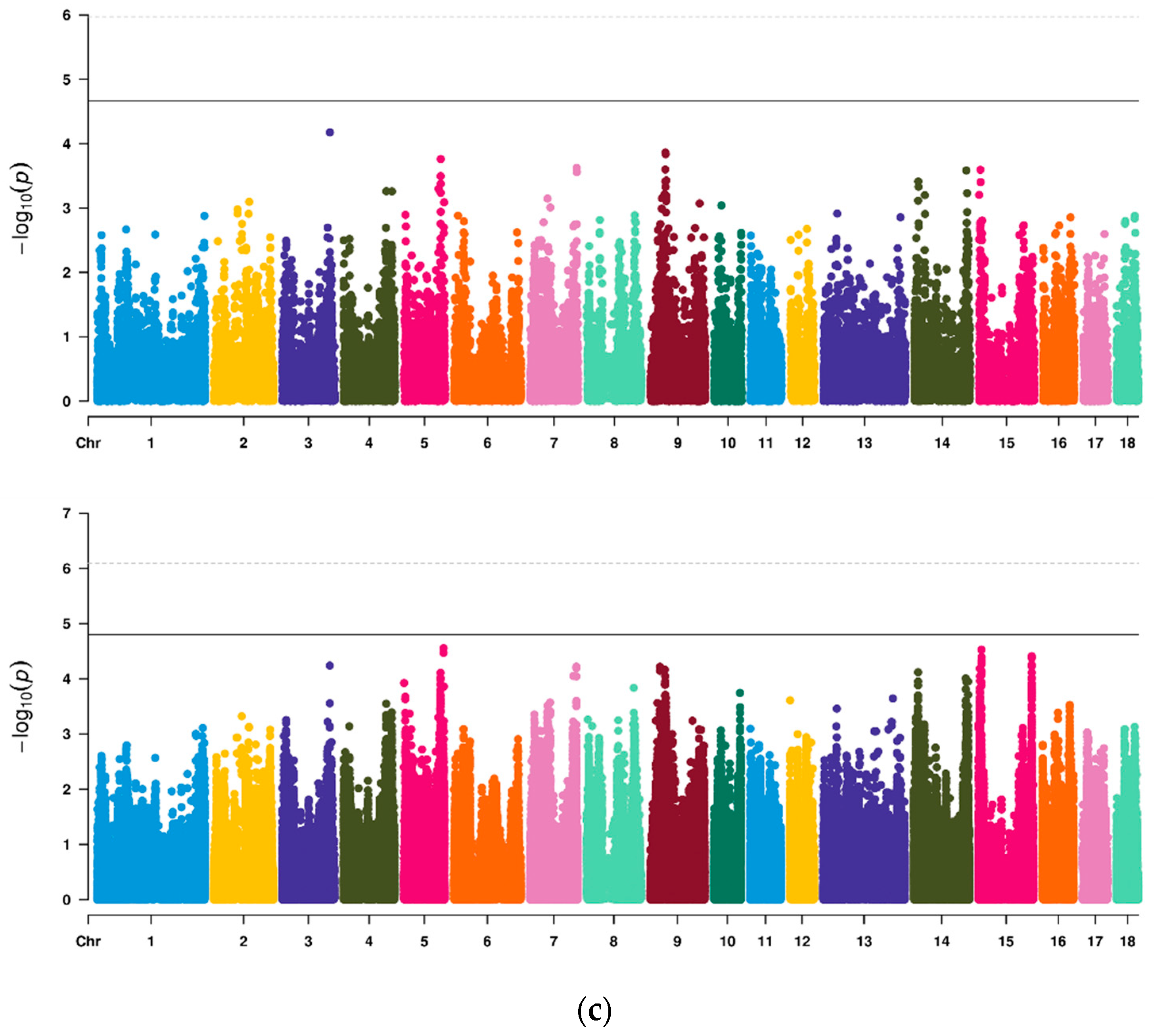
3.4. GWAS of Vulvar Traits in Suhuai Pigs
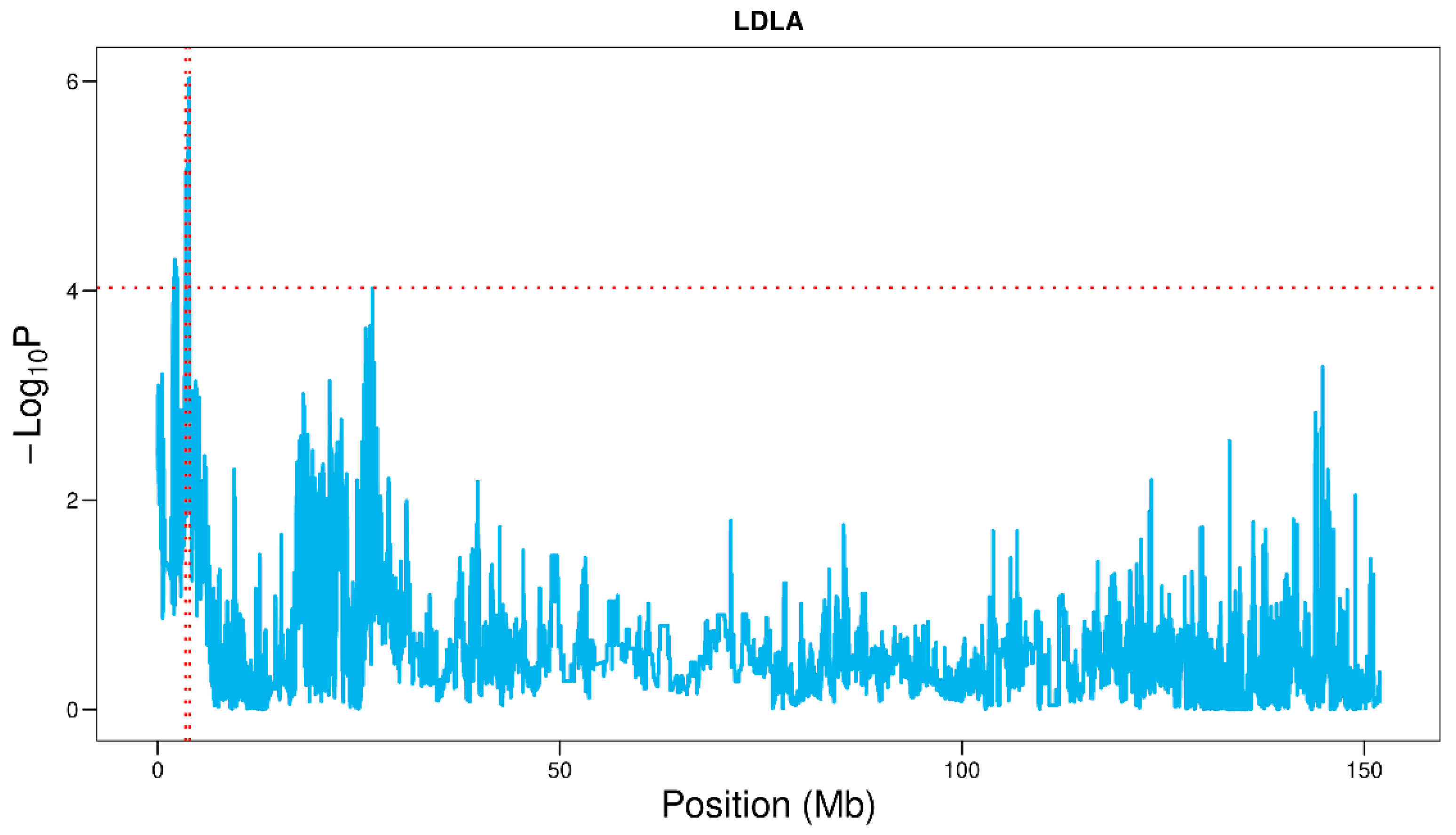
3.5. LDLA of Vulvar Traits in Suhuai Pigs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Graves, K.L.; Mordhorst, B.R.; Wright, E.C.; Hale, B.J.; Stalder, K.J.; Keating, A.F.; Ross, J.W. Identification of measures predictive of age of puberty onset in gilts. Transl. Anim. Sci. 2020, 4, 285–292. [Google Scholar] [CrossRef]
- Corredor, F.A.; Sanglard, L.P.; Ross, J.W.; Keating, A.F.; Leach, R.J.; Serão, N.V.L. Phenotypic and genomic relationships between vulva score categories and reproductive performance in first-parity sows. J. Anim. Sci. Biotechnol. 2021, 12, 7. [Google Scholar] [CrossRef]
- Ji, J.; Zhou, L.; Guo, Y.; Huang, L.; Ma, J. Genome-wide association study identifies 22 new loci for body dimension and body weight traits in a White Duroc×Erhualian F(2) intercross population. Asian-Australas J. Anim. Sci. 2017, 30, 1066–1073. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Guo, X.; Zhu, D.; Tan, C.; Bian, C.; Ren, J.; Huang, Z.; Zhao, Y.; Cai, G.; Liu, D.; et al. Accelerated deciphering of the genetic architecture of agricultural economic traits in pigs using a low-coverage whole-genome sequencing strategy. Gigascience 2021, 10, giab048. [Google Scholar] [CrossRef]
- Jiang, Y.; Tang, S.; Xiao, W.; Yun, P.; Ding, X. A genome-wide association study of reproduction traits in four pig populations with different genetic backgrounds. Asian-Australas J. Anim. Sci. 2020, 33, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Gòdia, M.; Reverter, A.; González-Prendes, R.; Ramayo-Caldas, Y.; Castelló, A.; Rodríguez-Gil, J.E.; Sánchez, A.; Clop, A. A systems biology framework integrating GWAS and RNA-seq to shed light on the molecular basis of sperm quality in swine. Genet. Sel. Evol. 2020, 52, 72. [Google Scholar] [CrossRef]
- Corredor, F.A.; Sanglard, L.P.; Leach, R.J.; Ross, J.W.; Keating, A.F.; Serão, N.V.L. Genetic and genomic characterization of vulva size traits in Yorkshire and Landrace gilts. BMC Genet. 2020, 21, 28. [Google Scholar] [CrossRef]
- Flossmann, G.; Wurmser, C.; Pausch, H.; Tenghe, A.; Dodenhoff, J.; Dahinten, G.; Götz, K.U.; Russ, I.; Fries, R. A nonsense mutation of bone morphogenetic protein-15 (BMP15) causes both infertility and increased litter size in pigs. BMC Genom. 2021, 22, 38. [Google Scholar] [CrossRef]
- Wang, B.; Li, P.; Zhou, W.; Gao, C.; Liu, H.; Li, H.; Niu, P.; Zhang, Z.; Li, Q.; Zhou, J.; et al. Association of Twelve Candidate Gene Polymorphisms with the Intramuscular Fat Content and Average Backfat Thickness of Chinese Suhuai Pigs. Animals 2019, 9, 858. [Google Scholar] [CrossRef] [Green Version]
- Mills, K.M.; Schinckel, A.P.; Stevens, J.G.; Casey, T.M.; Stewart, K.R. Evaluation of on-farm indicators of gilt reproductive performance potential at 21 days of age. Transl. Anim. Sci. 2020, 4, txaa210. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, B.L.; Browning, S.R. Genotype Imputation with Millions of Reference Samples. Am. J. Hum. Genet. 2016, 98, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Christensen, O.F.; Ostersen, T.; Wang, Y.; Lund, M.S.; Su, G. Improving genetic evaluation of litter size and piglet mortality for both genotyped and nongenotyped individuals using a single-step method. J. Anim. Sci. 2015, 93, 503–512. [Google Scholar] [CrossRef]
- Madsen, P.; Jensen, J.; Labouriau, R.; Christensen, O.F.; Sahana, G. DMU-a package for analyzing multivariate mixed models in quantitative genetics and genomics. In Proceedings of the 10th World Congress of Genetics Applied to Livestock Production, Vancouver, BC, Canada, 17–22 August 2014; pp. 18–22. [Google Scholar]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Speed, D.; Hemani, G.; Johnson, M.R.; Balding, D.J. Improved heritability estimation from genome-wide SNPs. Am. J. Hum. Genet. 2012, 91, 1011–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.B.; Hou, L.M.; Zhou, W.D.; Liu, H.; Tao, W.; Wu, W.J.; Niu, P.P.; Zhang, Z.P.; Zhou, J.; Li, Q.; et al. Genome-wide association study reveals a quantitative trait locus and two candidate genes on Sus scrofa chromosome 5 affecting intramuscular fat content in Suhuai pigs. Animal 2021, 15, 100341. [Google Scholar] [CrossRef]
- Schmid, M.; Maushammer, M.; Preuß, S.; Bennewitz, J. Mapping QTL for production traits in segregating Piétrain pig populations using genome-wide association study results of F2 crosses. Animal Genet. 2018, 49, 317–320. [Google Scholar] [CrossRef]
- Yang, Q.; Cui, J.; Chazaro, I.; Cupples, L.A.; Demissie, S. Power and type I error rate of false discovery rate approaches in genome-wide association studies. BMC Genet. 2005, 6 (Suppl. S1), S134. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Starmer, J.; Martin, E.R. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 2008, 32, 361–369. [Google Scholar] [CrossRef]
- Ye, S.; Chen, Z.T.; Zheng, R.; Diao, S.; Teng, J.; Yuan, X.; Zhang, H.; Chen, Z.; Zhang, X.; Li, J.; et al. New Insights From Imputed Whole-Genome Sequence-Based Genome-Wide Association Analysis and Transcriptome Analysis: The Genetic Mechanisms Underlying Residual Feed Intake in Chickens. Front. Genet. 2020, 11, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated tool for Genome-Wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Karim, L.; Takeda, H.; Lin, L.; Druet, T.; Arias, J.A.; Baurain, D.; Cambisano, N.; Davis, S.R.; Farnir, F.; Grisart, B.; et al. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat. Genet. 2011, 43, 405–413. [Google Scholar] [CrossRef]
- Xu, W.; Chen, D.; Yan, G.; Xiao, S.; Huang, T.; Zhang, Z.; Huang, L. Rediscover and Refine QTLs for Pig Scrotal Hernia by Increasing a Specially Designed F(3) Population and Using Whole-Genome Sequence Imputation Technology. Front. Genet. 2019, 10, 890. [Google Scholar] [CrossRef]
- Zhang, T.; Gao, H.; Sahana, G.; Zan, Y.; Fan, H.; Liu, J.; Shi, L.; Wang, H.; Du, L.; Wang, L.; et al. Genome-wide association studies revealed candidate genes for tail fat deposition and body size in the Hulun Buir sheep. J. Anim. Breed. Genet. 2019, 136, 362–370. [Google Scholar] [CrossRef]
- Yan, G.; Guo, T.; Xiao, S.; Zhang, F.; Xin, W.; Huang, T.; Xu, W.; Li, Y.; Zhang, Z.; Huang, L. Imputation-Based Whole-Genome Sequence Association Study Reveals Constant and Novel Loci for Hematological Traits in a Large-Scale Swine F(2) Resource Population. Front. Genet. 2018, 9, 401. [Google Scholar] [CrossRef]
- Knauer, M.T.; Cassady, J.P.; Newcom, D.W.; See, M.T. Estimates of variance components for genetic correlations among swine estrus traits. J. Anim. Sci. 2010, 88, 2913–2919. [Google Scholar] [CrossRef]
- Garcia-Gámez, E.; Gutiérrez-Gil, B.; Suarez-Vega, A.; de la Fuente, L.F.; Arranz, J.J. Identification of quantitative trait loci underlying milk traits in Spanish dairy sheep using linkage plus combined linkage disequilibrium and linkage analysis approaches. J. Dairy Sci. 2013, 96, 6059–6069. [Google Scholar] [CrossRef] [Green Version]
- Hirooka, H.; de Koning, D.J.; Harlizius, B.; van Arendonk, J.A.; Rattink, A.P.; Groenen, M.A.; Brascamp, E.W.; Bovenhuis, H. A whole-genome scan for quantitative trait loci affecting teat number in pigs. J. Anim. Sci. 2001, 79, 2320–2326. [Google Scholar] [CrossRef] [Green Version]
- Estienne, A.; Price, C.A. The fibroblast growth factor 8 family in the female reproductive tract. Reproduction 2018, 155, R53–R62. [Google Scholar] [CrossRef] [Green Version]
- Kir, S.; Kliewer, S.A.; Mangelsdorf, D.J. Roles of FGF19 in liver metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Choschzick, M.; Hess, S.; Tennstedt, P.; Holst, F.; Bohlken, H.; Gieseking, F.; Mahner, S.; Woelber, L.; Simon, R.; Sauter, G. Role of cyclin D1 amplification and expression in vulvar carcinomas. Hum. Pathol. 2012, 43, 1386–1393. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Traits | Number | Season | Batch |
|---|---|---|---|
| VL (cm) | 270 | ** | ** |
| VW (cm) | 270 | ** | * |
| VA (°) | 258 | * | non |
| Traits | Number | Max | Min | Mean | SE | CV (%) | h2 (SE) |
|---|---|---|---|---|---|---|---|
| VL (cm) | 270 | 5.7 | 1.2 | 3.40 | 0.05 | 25.46 | 0.23 (0.13) |
| VW (cm) | 270 | 4.3 | 0.9 | 2.62 | 0.04 | 27.67 | 0.32 (0.13) |
| VA (°) | 258 | 180 | 91 | 152.42 | 1.07 | 11.24 | 0.21 (0.12) |
| Trait | SSC | SNPs | Position | p-Value | Allele | MAF | PVE |
|---|---|---|---|---|---|---|---|
| VL | 2 | rs81344397 | 3,945,248 | 9.10 × 10−8 | C/T | 0.36 | 6.53% |
| 2 | rs81323795 | 3,726,771 | 1.05 × 10−7 | A/G | 0.36 | 6.47% | |
| 2 | rs319327579 | 3,747,849 | 1.52 × 10−7 | A/G | 0.36 | 6.33% | |
| 2 | rs336379732 | 3,831,753 | 2.13 × 10−7 | T/C | 0.36 | 6.22% | |
| 9 | rs326835497 | 118,063,093 | 1.71 × 10−5 | A/G | 0.27 | 5.09% | |
| VW | 2 | rs81323795 | 3,726,771 | 8.13 × 10−6 | A/G | 0.36 | 5.45% |
| 2 | rs319327579 | 3,747,849 | 9.39 × 10−6 | A/G | 0.36 | 5.36% | |
| 7 | rs80898557 | 11,989,354 | 2.04 × 10−5 | T/C | 0.43 | 4.33% | |
| 13 | rs320033947 | 21,085,416 | 1.31 × 10−5 | G/T | 0.43 | 8.19% |
| Traits | SSC | QTL Region (Mb) | Position of Lead SNP (bp) | p-Value of Lead SNP | PVE |
|---|---|---|---|---|---|
| VL | 2 | 3.25–4.25 | 3,833,112 | 6.98 × 10−8 | 6.71% |
| 7 | 7.23–8.07 | 7,763,002 | 7.74 × 10−7 | 5.27% | |
| 9 | 123.25–123.87 | 123,550,278 | 3.33 × 10−6 | 4.81% | |
| 16 | 12.66–13.26 | 12,961,112 | 1.52 × 10−5 | 4.94% | |
| 17 | 40.60–41.20 | 40,902,044 | 7.82 × 10−6 | 4.55% | |
| 17 | 43.10–43.72 | 43,404,836 | 1.06 × 10−5 | 3.94% | |
| VW | 1 | 271.53–272.13 | 271,827,161 | 1.37 × 10−5 | 4.99% |
| 2 | 3.41–4.05 | 3,726,718 | 4.40 × 10−6 | 5.81% | |
| 8 | 22.30–22.93 | 22,602,662 | 1.34 × 10−5 | 5.10% | |
| 11 | 7.29–8.10 | 7,650,870 | 5.19 × 10−6 | 5.04% | |
| 13 | 20.78–21.38 | 21,080,852 | 1.37 × 10−5 | 7.38% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, Y.; Hou, L.; Liu, C.; Li, K.; Guo, H.; Niu, P.; Li, Q.; Huang, R.; Li, P. Genome-Wide Association Study Identified a Quantitative Trait Locus and Two Candidate Genes on Sus scrofa Chromosome 2 Affecting Vulvar Traits of Suhuai Pigs. Genes 2022, 13, 1294. https://doi.org/10.3390/genes13081294
Yin Y, Hou L, Liu C, Li K, Guo H, Niu P, Li Q, Huang R, Li P. Genome-Wide Association Study Identified a Quantitative Trait Locus and Two Candidate Genes on Sus scrofa Chromosome 2 Affecting Vulvar Traits of Suhuai Pigs. Genes. 2022; 13(8):1294. https://doi.org/10.3390/genes13081294
Chicago/Turabian StyleYin, Yanzhen, Liming Hou, Chenxi Liu, Kaijun Li, Hao Guo, Peipei Niu, Qiang Li, Ruihua Huang, and Pinghua Li. 2022. "Genome-Wide Association Study Identified a Quantitative Trait Locus and Two Candidate Genes on Sus scrofa Chromosome 2 Affecting Vulvar Traits of Suhuai Pigs" Genes 13, no. 8: 1294. https://doi.org/10.3390/genes13081294
APA StyleYin, Y., Hou, L., Liu, C., Li, K., Guo, H., Niu, P., Li, Q., Huang, R., & Li, P. (2022). Genome-Wide Association Study Identified a Quantitative Trait Locus and Two Candidate Genes on Sus scrofa Chromosome 2 Affecting Vulvar Traits of Suhuai Pigs. Genes, 13(8), 1294. https://doi.org/10.3390/genes13081294