
Nucleosome-Omics: A Perspective on the Epigenetic Code and 3D Genome Landscape



Abstract
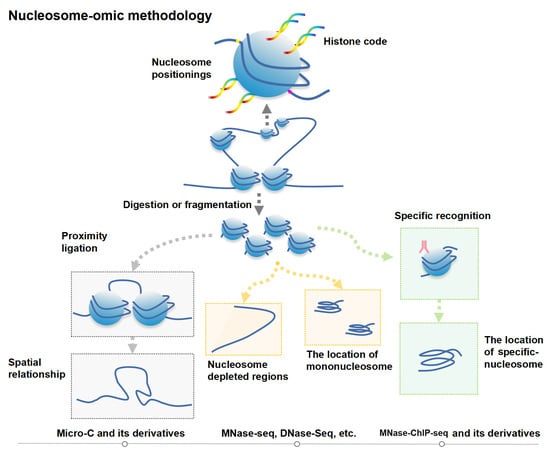
1. Introduction
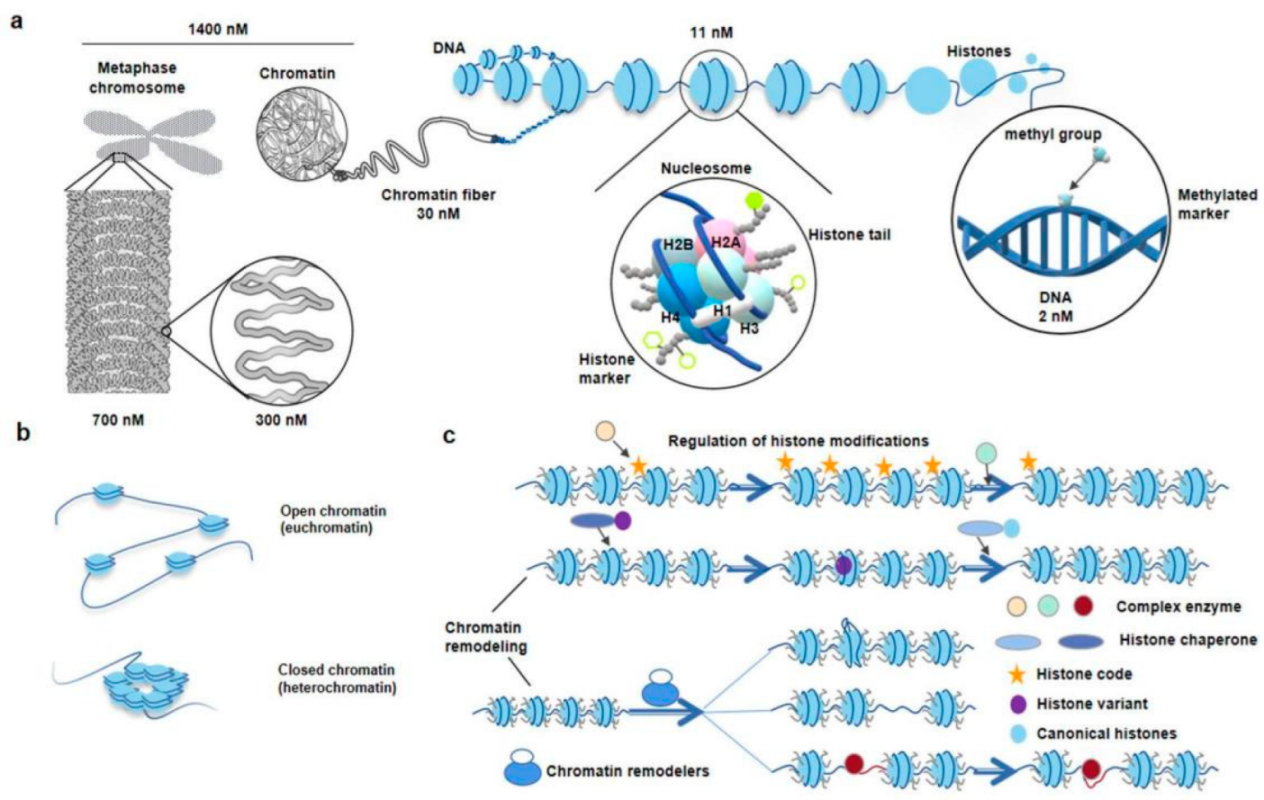
2. Nucleosome
2.1. Scientific Discovery, Development and Related Research of Nucleosomes
2.2. Significance of Studying Chromatin Nucleosome and Nucleosome-Level DNA
3. Genomics Techniques and Progress for Studying Epigenetic Phenomena at the Nucleosome Level
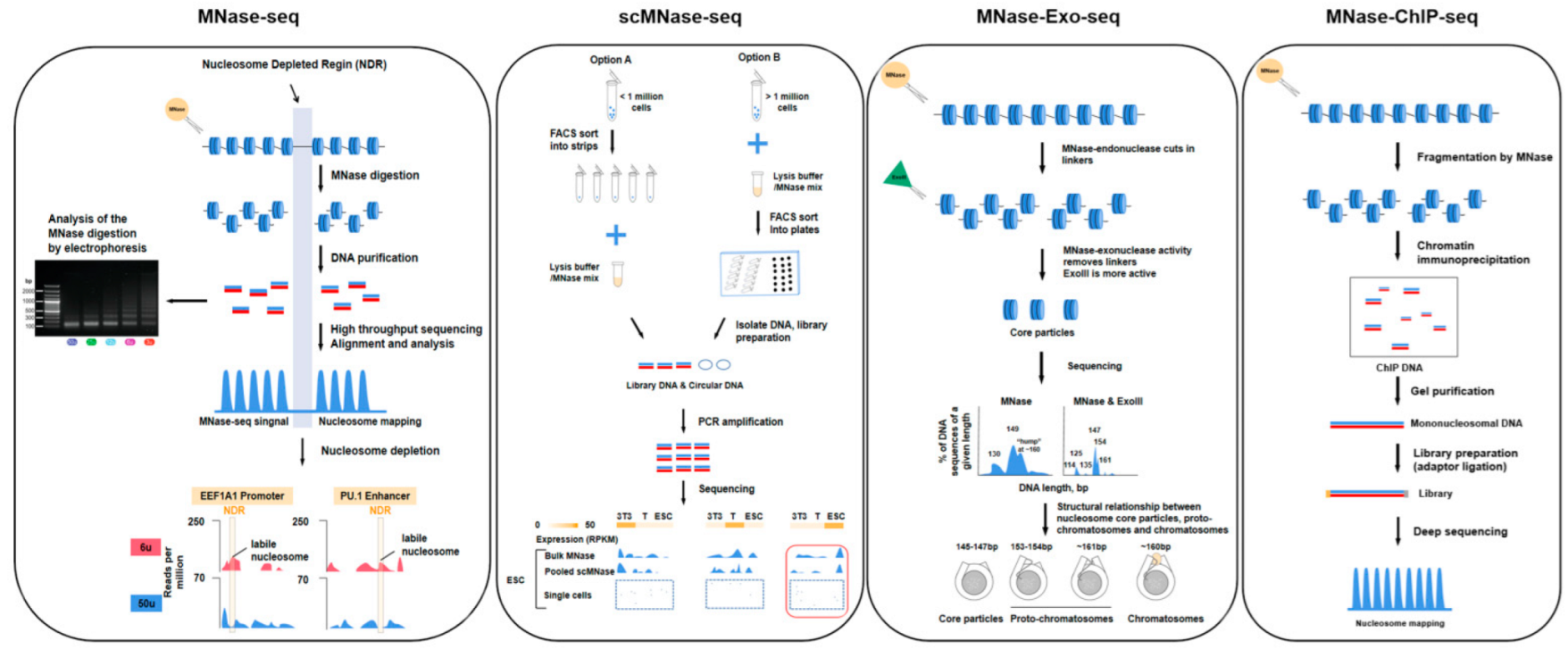
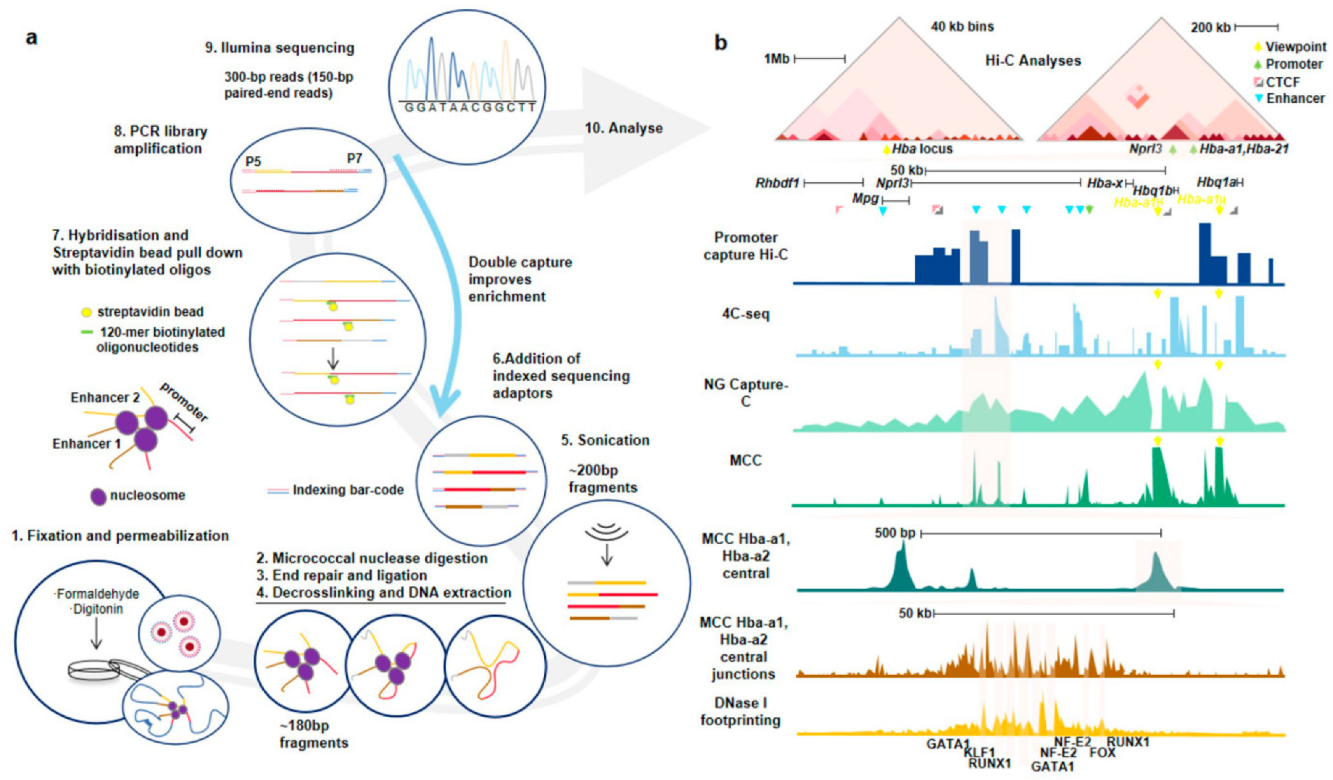
3.1. Nucleosome-Level Genomics Technology and Related Research
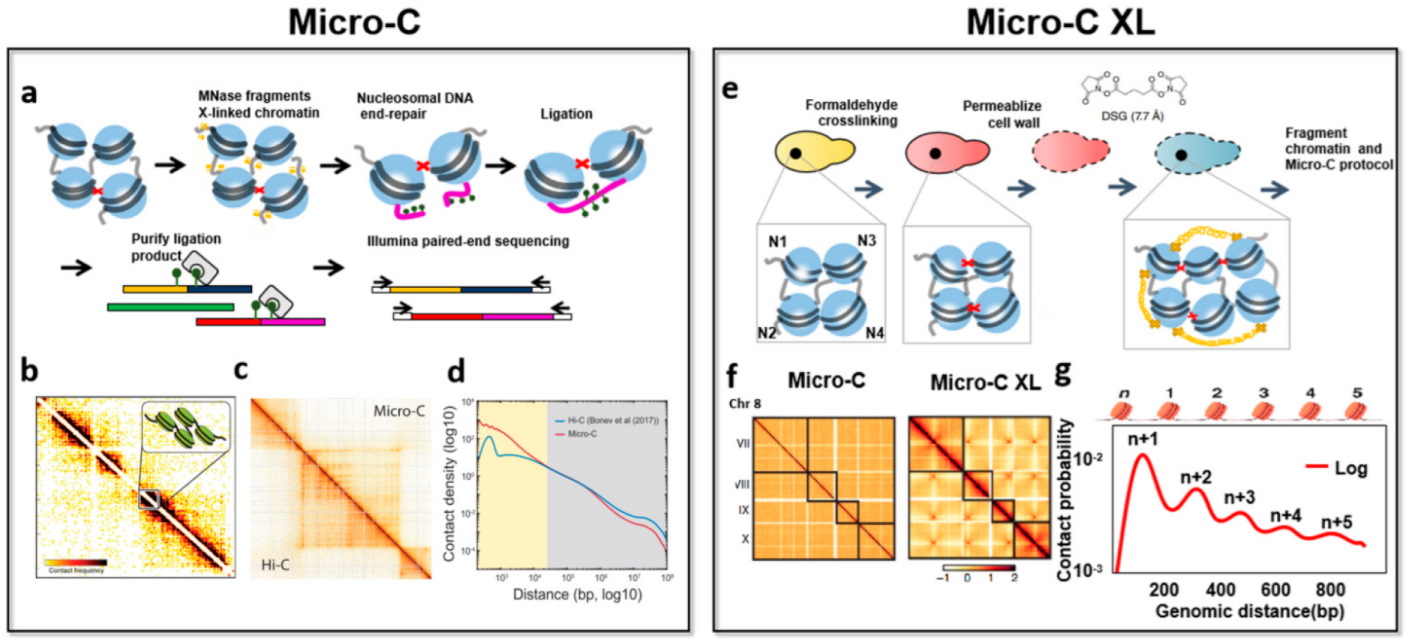
3.2. Nucleosome-Level 3D-Genomics Technology and Application Discovery
3.3. Comparing Nucleosome-Level Omics Techniques with Other Genomics Techniques
4. Research and Development Progress on Bioinformatics Analysis Tools and Pipelines for Nucleosomics
5. Conclusions and Future Perspectives of Nucleosome-Level Genomics Technologies and Applications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maloy, S.R.; Hughes, K.T. Brenner’s Encyclopedia of Genetics, 2nd ed.; Elsevier: Amsterdam, The Netherlands; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Goel, V.Y.; Hansen, A.S. The Macro and Micro of Chromosome Conformation Capture. Wiley Interdiscip. Rev. Dev. Biol. 2020, 10, e395. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Priest, D.; Taniguchi, Y. Nucleosome-Level 3D Organization of the Genome. Biochem. Soc. Trans. 2018, 46, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Oudet, P.; Gross-Bellard, M.; Chambon, P. Electron Microscopic and Biochemical Evidence That Chromatin Structure Is A Repeating Unit. Cell 1975, 4, 281–300. [Google Scholar] [CrossRef]
- Olins, A.L.; Olins, D.E. Spheroid Chromatin Units (V Bodies). Science 1974, 183, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Finch, J.T.; Klug, A. Solenoidal Model for Superstructure in Chromatin. Proc. Natl. Acad. Sci. USA 1976, 73, 1897–1901. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, C.L.; Frado, L.L.; Rattner, J.B. The Higher-Order Structure of Chromatin: Evidence for a Helical Ribbon Arrangement. J. Cell Biol. 1984, 99, 42–52. [Google Scholar] [CrossRef]
- Williams, S.; Athey, B.; Muglia, L.; Schappe, R.; Gough, A.; Langmore, J. Chromatin Fibers are Left-Handed Double Helices with Diameter and Mass Per Unit Length That Depend on Linker Length. Biophys. J. 1986, 49, 233–248. [Google Scholar] [CrossRef]
- Eltsov, M.; MacLellan, K.M.; Maeshima, K.; Frangakis, A.S.; Dubochet, J. Analysis of Cryo-Electron Microscopy Images Does Not Support the Existence of 30-Nm Chromatin Fibers In Mitotic Chromosomes In Situ. Proc. Natl. Acad. Sci. USA 2008, 105, 19732–19737. [Google Scholar] [CrossRef]
- Song, F.; Chen, P.; Sun, D.; Wang, M.; Dong, L.; Liang, D.; Xu, R.-M.; Zhu, P.; Li, G. Cryo-EM Study of the Chromatin Fiber Reveals a Double Helix Twisted by Tetranucleosomal Units. Science 2014, 344, 376–380. [Google Scholar] [CrossRef]
- Schwartz, U.; Németh, A.; Diermeier, S.; Exler, J.H.; Hansch, S.; Maldonado, R.; Heizinger, L.; Merkl, R.; Längst, G. Characterizing the Nuclease Accessibility of DNA in Human Cells to Map Higher Order Structures of Chromatin. Nucleic Acids Res. 2019, 47, 1239–1254. [Google Scholar] [CrossRef]
- Ocampo, J.; Cui, F.; Zhurkin, V.B.; Clark, D.J. The Proto-Chromatosome: A Fundamental Subunit of Chromatin? Nucleus 2016, 7, 382–387. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Z.; Pugh, B.F. High-Resolution Genome-wide Mapping of the Primary Structure of Chromatin. Cell 2011, 144, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Mieczkowski, J.; Cook, A.; Bowman, S.K.; Mueller, B.; Alver, B.H.; Kundu, S.; Deaton, A.M.; Urban, J.A.; Larschan, E.; Park, P.J.; et al. MNase Titration Reveals Differences Between Nucleosome Occupancy And Chromatin Accessibility. Nat. Commun. 2016, 7, 11485. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xiong, C.; Zhuo, B.; Wen, Z.; Shen, J.; Liu, C.; Chang, L.; Wang, K.; Wang, M.; Wu, C.; et al. Analysis of Local Chromatin States Reveals Gene Transcription Potential During Mouse Neural Progenitor Cell Differentiation. Cell Rep. 2020, 32, 108072. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Zhang, Y. Deciphering Hi-C: From 3D genome to function. Cell Biol. Toxicol. 2019, 35, 15–32. [Google Scholar] [CrossRef]
- Hsieh, T.-H.S.; Weiner, A.; Lajoie, B.; Dekker, J.; Friedman, N.; Rando, O.J. Mapping Nucleosome Resolution Chromosome Folding in Yeast by Micro-C. Cell 2015, 162, 108–119. [Google Scholar] [CrossRef]
- Mozziconacci, J.; Koszul, R. Filling the Gap: Micro-C Accesses the Nucleosomal Fiber at 100-1000 Bp Resolution. Genome Biol. 2015, 16, 169. [Google Scholar] [CrossRef]
- Chang, L.-H.; Noordermeer, D. Of Dots and Stripes: The Morse Code of Micro-C Reveals the Ultrastructure of Transcriptional and Architectural Mammalian 3D Genome Organization. Mol. Cell 2020, 78, 376–378. [Google Scholar] [CrossRef]
- Ou, H.D.; Phan, S.; Deerinck, T.J.; Thor, A.; Ellisman, M.H.; O’Shea, C.C. ChromEMT: Visualizing 3D Chromatin Structure and Compaction in Interphase and Mitotic Cells. Science 2017, 357, eaag0025. [Google Scholar] [CrossRef]
- Kornberg, R.D.; Thomas, J.O. Chromatin Structure: Oligomers of the Histones. Science 1974, 184, 865–868. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal Structure of the Nucleosome Core Particle at 2.8 a Resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Barbier, J.; Vaillant, C.; Volff, J.N.; Brunet, F.G.; Audit, B. Coupling between Sequence-Mediated Nucleosome Organization and Genome Evolution. Genes 2021, 12, 851. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S. Nucleosome Destabilization in the Epigenetic Regulation of Gene Expression. Nat. Rev. Genet. 2008, 9, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fan, X.; Yan, J.; Chen, M.; Zhu, M.; Tang, Y.; Liu, S.; Tang, Z. A Comprehensive Epigenome Atlas Reveals DNA Methylation Regulating Skeletal Muscle Development. Nucleic Acids Res. 2021, 49, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, C.; Liu, W.; Li, J.; Li, C.; Kou, X.; Chen, J.; Zhao, Y.; Gao, H.; Wang, H.; et al. Distinct Features of H3K4me3 and H3K27me3 Chromatin Domains in Pre-Implantation Embryos. Nature 2016, 537, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K.; Segal, E. Determinants of Nucleosome Positioning. Nat. Struct. Mol. Biol. 2013, 20, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Davey, C.A. Nucleosome Structural Studies. Curr. Opin. Struct. Biol. 2011, 21, 128–136. [Google Scholar] [CrossRef]
- Hoboth, P.; Sztacho, M.; Šebesta, O.; Schätz, M.; Castano, E.; Hozák, P. Nanoscale Mapping of Nuclear Phosphatidylinositol Phosphate Landscape by Dual-Color dSTORM. Biochim. et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2021, 1866, 158890. [Google Scholar] [CrossRef]
- Sztacho, M.; Salovska, B.; Cervenka, J.; Balaban, C.; Hoboth, P.; Hozak, P. Limited Proteolysis-Coupled Mass Spectrometry Identifies Phosphatidylinositol 4,5-Bisphosphate Effectors in Human Nuclear Proteome. Cells 2021, 10, 68. [Google Scholar] [CrossRef]
- Sharma, A.B.; Dimitrov, S.; Hamiche, A.; Van Dyck, E. Centromeric and Ectopic Assembly of CENP-A Chromatin in Health and Cancer: Old Marks and New Tracks. Nucleic Acids Res. 2019, 47, 1051–1069. [Google Scholar] [CrossRef]
- Zhou, C.Y.; Johnson, S.L.; Gamarra, N.I.; Narlikar, G.J. Mechanisms of ATP-Dependent Chromatin Remodeling Motors. Annu. Rev. Biophys. 2016, 45, 153–181. [Google Scholar] [CrossRef] [PubMed]
- Eitoku, M.; Sato, L.; Senda, T.; Horikoshi, M. Histone Chaperones: 30 years from Isolation to Elucidation of the Mechanisms of Nucleosome Assembly and Disassembly. Cell. Mol. Life Sci. 2007, 65, 414–444. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. The Molecular Basis of Eucaryotic Transcription. Cell Death Differ. 2008, 14, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zheng, W.; Jiang, Y.; Ding, G.; Hou, X.; Tang, Y.; Li, Y.; Gao, S.; Chang, G.; Zhang, X.; et al. Nucleosome Organizations in Induced Pluripotent Stem Cells Reprogrammed from Somatic Cells Belonging to Three Different Germ Layers. BMC Biol. 2014, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Schones, D.E.; Cui, K.; Cuddapah, S.; Roh, T.-Y.; Barski, A.; Wang, Z.; Wei, G.; Zhao, K. Dynamic Regulation of Nucleosome Positioning in the Human Genome. Cell 2008, 132, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Lohr, D.; Kovacic, R.T.; Van Holde, K.E. Quantitative Analysis of the Digestion of Yeast Chromatin by Staphylococcal Nuclease. Biochemistry 1977, 16, 463–471. [Google Scholar] [CrossRef]
- Pajoro, A.; Muiño, J.M.; Angenent, G.C.; Kaufmann, K. Profiling Nucleosome Occupancy by MNase-seq: Experimental Protocol and Computational Analysis. Methods Mol. Biol. 2018, 1675, 167–181. [Google Scholar] [CrossRef]
- Noll, M. Subunit Structure of Chromatin. Nature 1974, 251, 249–251. [Google Scholar] [CrossRef]
- Gao, W.; Lai, B.; Ni, B.; Zhao, K. Genome-Wide Profiling of Nucleosome Position and Chromatin Accessibility in Single Cells Using ScMnase-Seq. Nat. Protoc. 2020, 15, 68–85. [Google Scholar] [CrossRef]
- Wang, C.; Chen, C.; Liu, X.; Li, C.; Wu, Q.; Chen, X.; Yang, L.; Kou, X.; Zhao, Y.; Wang, H.; et al. Dynamic Nucleosome Organization After Fertilization Reveals Regulatory Factors for Mouse Zygotic Genome Activation. Cell Res. 2022, in press. [Google Scholar] [CrossRef]
- Liu, Y.; Lay, F.D.; Liang, G.; Berman, B.P.; Jones, P.A.; Kelly, T. Genome-wide Mapping of Nucleosome Positioning and DNA Methylation Within Individual DNA Molecules. J. Biomol. Tech. 2012, 23, S4. [Google Scholar]
- Zhao, H.; Zhang, W.; Zhang, T.; Lin, Y.; Hu, Y.; Fang, C.; Jiang, J. Genome-Wide Mnase Hypersensitivity Assay Unveils Distinct Classes of Open Chromatin Associated with H3k27me3 and DNA Methylation in Arabidopsis thaliana. Genome Biol. 2020, 21, 24. [Google Scholar] [CrossRef] [PubMed]
- Baldi, S.; Krebs, S.; Blum, H.; Becker, P.B. Genome-Wide Measurement of Local Nucleosome Array Regularity and Spacing by Nanopore Sequencing. Nat. Struct. Mol. Biol. 2018, 25, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Ahmad, K.; Henikoff, S. Transcription and Remodeling Produce Asymmetrically Unwrapped Nucleosomal Intermediates. Mol. Cell 2017, 68, 1038–1053.e4. [Google Scholar] [CrossRef] [PubMed]
- Chereji, R.; Ocampo, J.; Clark, D.J. MNase-Sensitive Complexes in Yeast: Nucleosomes and Non-histone Barriers. Mol. Cell 2017, 65, 565–577.e3. [Google Scholar] [CrossRef]
- Montanera, K.N.; Rhee, H.S. High-Resolution Mapping of Protein-DNA Interactions in Mouse Stem Cell-Derived Neurons Using Chromatin Immunoprecipitation-Exonuclease (Chip-Exo). JoVE 2020, 162, 61124. [Google Scholar] [CrossRef]
- Han, G.C.; Vinayachandran, V.; Bataille, A.R.; Park, B.; Chan-Salis, K.Y.; Keller, C.A.; Long, M.; Mahony, S.; Hardison, R.; Pugh, B.F. Genome-Wide Organization of GATA1 and TAL1 Determined at High Resolution. Mol. Cell. Biol. 2016, 36, 157–172. [Google Scholar] [CrossRef]
- Lion, M.; Muhire, B.; Namiki, Y.; Tolstorukov, M.Y.; Oettinger, M.A. Alterations in Chromatin at Antigen Receptor Loci Define Lineage Progression During B Lymphopoiesis. Proc. Natl. Acad. Sci. USA 2020, 117, 5453–5462. [Google Scholar] [CrossRef]
- Esnault, C.; Magat, T.; García-Oliver, E.; Andrau, J.C. Analyses of Promoter, Enhancer, and Nucleosome Organization in Mammalian Cells by Mnase-Seq. Methods Mol. Biol. 2021, 2351, 93–104. [Google Scholar]
- Lai, B.; Gao, W.; Cui, K.; Xie, W.; Tang, Q.; Jin, W.; Hu, G.; Ni, B.; Zhao, K. Principles of Nucleosome Organization Revealed by Single-Cell Micrococcal Nuclease Sequencing. Nature 2018, 562, 281–285. [Google Scholar] [CrossRef]
- Wal, M.; Pugh, B.F. Genome-Wide Mapping of Nucleosome Positions in Yeast Using High-Resolution Mnase Chip-Seq. Methods Enzymol. 2012, 513, 233–250. [Google Scholar] [PubMed]
- Uzun, I.; Brown, T.; Fischl, H.; Angel, A.; Mellor, J. Spt4 Facilitates the Movement of RNA Polymerase II Through the +2 Nucleosomal Barrier. Cell Rep. 2021, 36, 109755. [Google Scholar] [CrossRef] [PubMed]
- de Souza, N. Genomics. Micro-C Maps of Genome Structure. Nat. Methods 2015, 12, 812. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.-H.S.; Fudenberg, G.; Goloborodko, A.; Rando, O.J. Micro-C XL: Assaying Chromosome Conformation from the Nucleosome to the Entire Genome. Nat. Methods 2016, 13, 1009–1011. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, Z.; Yang, S.; Sun, G.; Jia, L.; Zheng, Z.; Gu, Q.; Tao, W.; Cheng, T.; Li, C.; et al. Reveals 3d Chromatin Architecture Dynamics During Mouse Hematopoiesis. Cell Rep. 2020, 32, 108206. [Google Scholar] [CrossRef]
- Chereji, R.V.; Bryson, T.D.; Henikoff, S. Quantitative MNase-seq Accurately Maps Nucleosome Occupancy Levels. Genome Biol. 2019, 20, 108206. [Google Scholar] [CrossRef]
- Deng, W.H.; Li, X.H. Resolving nucleosomal positioning and occupancy with MNase-seq. Hereditas 2020, 42, 1143–1155. [Google Scholar]
- Park, P.J. Chip-Seq: Advantages and Challenges of a Maturing Technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef]
- Rhee, H.S.; Pugh, B.F. Comprehensive Genome-Wide Protein-DNA Interactions Detected at Single-Nucleotide Resolution. Cell 2011, 147, 1408–1419. [Google Scholar] [CrossRef]
- van Essen, D.; Oruba, A.; Saccani, S. High-resolution Chip-Mnase Mapping of Nucleosome Positions at Selected Genomic Loci and Alleles. Methods Mol. Biol. 2021, 2351, 123–145. [Google Scholar]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. Atac-Seq: A method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 9. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Crawford, G.E. DNase-seq: A High-Resolution Technique for Mapping Active Gene Regulatory Elements Across the Genome from Mammalian Cells. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5384. [Google Scholar] [CrossRef] [PubMed]
- Rhie, S.K.; Schreiner, S.; Farnham, P.J. Defining Regulatory Elements in the Human Genome Using Nucleosome Occupancy and Methylome Sequencing (NOMe-Seq). Methods Mol. Biol. 2018, 1766, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.; Mieczkowski, J.; Tolstorukov, M.Y. Single-Assay Profiling of Nucleosome Occupancy and Chromatin Accessibility. Curr. Protoc. Mol. Biol. 2017, 120, 18. [Google Scholar] [CrossRef]
- Tao, S.; Lin, K.; Zhu, Q.; Zhang, W. MH-Seq for Functional Characterization of Open Chromatin in Plants. Trends Plant Sci. 2020, 25, 618–619. [Google Scholar] [CrossRef]
- Fouse, S.D.; Nagarajan, R.P.; Costello, J.F. Genome-Scale DNA Methylation Analysis. Epigenomics 2010, 2, 105–117. [Google Scholar] [CrossRef]
- Zheng, M.; Tian, S.Z.; Capurso, D.; Kim, M.; Maurya, R.; Lee, B.; Kim, M.; Capurso, D.; Piecuch, E.; Gong, L.; et al. Multiplex Chromatin Interaction Analysis with Single-Molecule Precision. bioRxiv 2018. [Google Scholar] [CrossRef]
- Krietenstein, N.; Abraham, S.; Venev, S.V.; Abdennur, N.; Gibcus, J.; Hsieh, T.-H.S.; Parsi, K.M.; Yang, L.; Maehr, R.; Mirny, L.A.; et al. Ultrastructural Details of Mammalian Chromosome Architecture. Mol. Cell 2020, 78, 554–565.e7. [Google Scholar] [CrossRef]
- Hsieh, T.S.; Cattoglio, C.; Slobodyanyuk, E.; Hansen, A.S.; Rando, O.J.; Tjian, R.; Darzacq, X. Resolving the 3D Landscape of Transcription-Linked Mammalian Chromatin Folding. Mol. Cell 2020, 78, 539–553. [Google Scholar] [CrossRef]
- Swygert, S.G.; Lin, D.; Portillo-Ledesma, S.; Lin, P.Y.; Hunt, D.R.; Kao, C.F.; Schlick, T.; Noble, W.S.; Tsukiyama, T. Local chromatin fiber folding represses transcription and loop extrusion in quiescent cells. ELife 2021, 10, e72062. [Google Scholar] [CrossRef]
- Boyle, A.P.; Davis, S.; Shulha, H.P.; Meltzer, P.; Margulies, E.H.; Weng, Z.; Furey, T.S.; Crawford, G.E. High-Resolution Mapping and Characterization of Open Chromatin Across the Genome. Cell 2008, 132, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Luo, K.; Winter, P.S.; Crawford, G.E.; Iversen, E.S.; Hartemink, A.J. Mapping Nucleosome Positions Using DNase-seq. Genome Res. 2016, 26, 351–364. [Google Scholar] [CrossRef]
- Rhie, S.; Yao, L.; Luo, Z.; Witt, H.; Schreiner, S.; Guo, Y.; Perez, A.; Farnham, P.J. ZFX Acts as a Transcriptional Activator in Multiple Types of Human Tumors by Binding Downstream from Transcription Start Sites at the Majority of CpG Island Promoters. Genome Res. 2018, 28, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Taberlay, P.C.; Statham, A.L.; Kelly, T.K.; Clark, S.J.; Jones, P.A. Reconfiguration of Nucleosome-Depleted Regions at Distal Regulatory Elements Accompanies DNA Methylation of Enhancers and Insulators in Cancer. Genome Res. 2014, 24, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Hua, P.; Badat, M.; Hanssen, L.L.P.; Hentges, L.D.; Crump, N.; Downes, D.J.; Jeziorska, D.M.; Oudelaar, A.M.; Schwessinger, R.; Taylor, S.; et al. Defining Genome Architecture at Base-Pair Resolution. Nature 2021, 595, 125–129. [Google Scholar] [CrossRef]
- Ohno, M.; Ando, T.; Priest, D.G.; Kumar, V.; Yoshida, Y.; Taniguchi, Y. Sub-nucleosomal Genome Structure Reveals Distinct Nucleosome Folding Motifs. Cell 2019, 176, 520–534.e25. [Google Scholar] [CrossRef]
- Teif, V.B. Nucleosome Positioning: Resources and Tools Online. Brief. Bioinform. 2016, 17, 745–757. [Google Scholar] [CrossRef]
- ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Kawaji, H.; Severin, J.; Lizio, M.; Waterhouse, A.; Katayama, S.; Irvine, K.M.; Hume, D.A.; Forrest, A.R.; Suzuki, H.; Carninci, P.; et al. The FANTOM Web Resource: From Mammalian Transcriptional Landscape to Its Dynamic Regulation. Genome Biol. 2009, 10, R40. [Google Scholar] [CrossRef]
- Kolmykov, S.; Yevshin, I.; Kulyashov, M.; Sharipov, R.; Kondrakhin, Y.; Makeev, V.J.; Kulakovskiy, I.V.; Kel, A.; Kolpakov, F. GTRD: An Integrated View of Transcription Regulation. Nucleic Acids Res. 2021, 49, D104–D111. [Google Scholar] [CrossRef]
- Feng, F.; Yao, Y.; Wang, X.Q.D.; Zhang, X.; Liu, J. Connecting High-Resolution 3d Chromatin Organization with Epigenomics. Nat. Commun. 2022, 13, 2054. [Google Scholar] [CrossRef]
- Fang, K.; Li, T.; Huang, Y.; Jin, V.X. Nuchmm: A Method for Quantitative Modeling of Nucleosome Organization Identifying Functional Nucleosome States Distinctly Associated with Splicing Potentiality. Genome Biol. 2021, 22, 250. [Google Scholar] [CrossRef]
- Sefer, E. ProbC: Joint Modeling of Epigenome and Transcriptome Effects in 3D Genome. BMC Genom. 2022, 23, 287. [Google Scholar] [CrossRef]
- Tran, T.Q.; MacAlpine, H.K.; Tripuraneni, V.; Mitra, S.; MacAlpine, D.M.; Hartemink, A.J. Linking the Dynamics of Chromatin Occupancy and Transcription with Predictive Models. Genome Res. 2021, 31, 1035–1046. [Google Scholar] [CrossRef]
- Levitsky, V.G.; Katokhin, A.V.; Podkolodnaya, O.A.; Furman, D.P.; Kolchanov, N.A. NPRD: Nucleosome Positioning Region Database. Nucleic Acids Res. 2004, 33, D67–D70. [Google Scholar] [CrossRef][Green Version]
- Oki, S.; Ohta, T.; Shioi, G.; Hatanaka, H.; Ogasawara, O.; Okuda, Y.; Kawaji, H.; Nakaki, R.; Sese, J.; Meno, C. Chip-Atlas: A Data-Mining Suite Powered by Full Integration of Public chip-seq data. EMBO Rep. 2018, 19, e46255. [Google Scholar] [CrossRef]
- Zheng, R.; Wan, C.; Mei, S.; Qin, Q.; Wu, Q.; Sun, H.; Chen, C.-H.; Brown, M.; Zhang, X.; Meyer, C.A.; et al. Cistrome Data Browser: Expanded Datasets and New Tools for Gene Regulatory Analysis. Nucleic Acids Res. 2018, 47, D729–D735. [Google Scholar] [CrossRef]
- Kazachenka, A.; Bertozzi, T.M.; Sjoberg-Herrera, M.K.; Walker, N.; Gardner, J.; Gunning, R.; Pahita, E.; Adams, S.; Adams, D.; Ferguson-Smith, A.-C. Identification, Characterization, and Heritability of Murine Metastable Epialleles: Implications for Non-Genetic Inheritance. Cell 2018, 175, 1717. [Google Scholar] [CrossRef]
- Yang, J.H.; Li, J.H.; Jiang, S.; Zhou, H.; Qu, L.H. Chipbase: A Database for Decoding the Transcriptional Regulation of Long Non-Coding Rna and Microrna Genes from Chip-Seq Data. Nucleic Acids Res. 2013, 41, D177–D187. [Google Scholar] [CrossRef]
- Chèneby, J.; Ménétrier, Z.; Mestdagh, M.; Rosnet, T.; Douida, A.; Rhalloussi, W.; Bergon, A.; Lopez, F.; Ballester, B. ReMap 2020: A Database of Regulatory Regions from an Integrative Analysis of Human and Arabidopsis DNA-Binding Sequencing Experiments. Nucleic Acids Res. 2019, 48, D180–D188. [Google Scholar] [CrossRef]
- Wang, J.; Zhuang, J.; Iyer, S.; Lin, X.-Y.; Greven, M.C.; Kim, B.-H.; Moore, J.; Pierce, B.; Dong, X.; Virgil, D.; et al. Factorbook.org: A Wiki-Based Database for Transcription Factor-Binding Data Generated by the ENCODE Consortium. Nucleic Acids Res. 2012, 41, D171–D176. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, J.; Liang, F.; Liu, Y.; Wang, Q.; Zhang, H.; Jiang, M.; Zhang, Z.; Zhao, W.; Bao, Y.; et al. NucMap: A Database of Genome-Wide Nucleosome Positioning Map Across Species. Nucleic Acids Res. 2018, 47, D163–D169. [Google Scholar] [CrossRef]
- Shtumpf, M.; Piroeva, K.V.; Agrawal, S.P.; Jacob, D.R.; Teif, V.B. NucPosDB: A Database of Nucleosome Positioning In Vivo and Nucleosomics of Cell-Free DNA. Chromosoma 2022, 131, 19–28. [Google Scholar] [CrossRef]
- Liu, Y.; Bisio, H.; Toner, C.M.; Jeudy, S.; Philippe, N.; Zhou, K.; Bowerman, S.; White, A.; Edwards, G.; Abergel, C.; et al. Virus-Encoded Histone Doublets Are Essential and Form Nucleosome-Like Structures. Cell 2021, 184, 4237–4250.e19. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technology | Sequence Type | Start Material | Enzyme Digestion | Advantage | Feature | Reference |
|---|---|---|---|---|---|---|
| MNase-seq | Single-end or paired-end sequencing | 10–20 million cells | MNase | The whole genome can be measured, high resolution, low technical difficulty. | MNase has sequence bias, it cuts DNA upstream of A or T faster than upstream of G or C, traditional method requires a large number of cells, easy to cause technical error. | [57,58] |
| scMNase-seq | Single-end or paired-end sequencing | >0.1 million cells | MNase | Provides a method for determining nucleosome localization and chromatin accessibility in single-cell or low-input materials, few cells are required, high resolution, low technical difficulty. | Low capture rate, information may be missing, lower throughput. | [40] |
| ULI-MNase-seq | Paired-end sequencing | 10–15 pronuclei per replicate | MNase | Low cell initiation. | It requires extremely high proficiency and skill. It easily loses cell and library DNA. | [41] |
| MNase-Exo-seq | Paired-end sequencing | 10–20 million cells | MNase, exonuclease III | The perfectly clipped core particle has a major sharp peak corresponding to it, weaker signals that cannot be detected in MNase-seq data are evident. | Detailed analysis of nucleosome location is complicated, it is possible to misestimate the occupancy of a specific nucleosome positions. | [12] |
| ChIP-seq | Single-end or paired-end sequencing | >1 million cells | Priority with MNase | High resolution and low noise, high genome coverage and wider dynamic range, the method is more mature and widely applicable. | Data quality depends on antibody quality, and antibody screening is time-consuming and costly. | [58,59] |
| ChIP-exo | Single-end or paired-end sequencing | >1 million cells | Exonuclease | High resolution, defines genomic binding locations, more precisely determine the location of protein gene interactions in the genome, few false positives or negatives Binding-site complexity. | Multiple binding of a single protein cannot be detected. | [60] |
| ChIP-MNase | Single-end or paired-end sequencing | 10–20 million cells | MNase | High resolution, nucleosome localization analysis in specific position of the genome and differential analysis of alleles undergoing different molecular processes. | Need for precise selection of antibodies, antibody repertoire may be incomplete, Other features similar to MNase-seq. | [61] |
| ATAC-seq | Paired-end sequencing | 500–50,000 cells | Tn5 transposase | Simple method, short experiment period, few cells are required, high resolution and good repeatability. | Conventional data analysis methods have limitations, Tn5 transposase is expensive. | [58,62] |
| DNase-seq | Single-end or paired-end sequencing | >1 million cells | DNase I | Simple method, high resolution, the most active regulatory regions can be identified from many cell types. | Traditional method requires a large number of cells, precise control of enzyme quantity, time consuming; it was not easy to determine the precise activity and function which were associated with each regulatory region. | [58,63] |
| NOMe-seq | Paired-end sequencing | Need to test | GpC methyltransferase | Does not depend on knowing the exact modification of surrounding nucleosomes. It can provide localization information of multiple nucleosomes on both sides of each open regulatory element, nucleosome localization and DNA methylation degree can be analyzed simultaneously. | Requires a large number of cells and data analysis is difficult. | [58,64] |
| Micro-C | Paired-end sequencing | 0.001–5 million cells | MNase | The signal-to-noise is improved, high resolution, reveals the chromosome folding of nucleosome resolution. | The observed chromosome structure will be biased, difficulty recovering known higher-order interactions. | [17] |
| Micro-C XL | Paired-end sequencing | 1 million cells | MNase | The signal-to-noise is improved, improved structure visualization, chromosome folding can be detected from nucleosomes to whole genomes, adds some subtle details to the Micro-C map. | Requires one more step of cross-linking, it may take many attempts to find the best conditions. | [55] |
| MACC-seq | Single-end or paired-end sequencing | 1 million cells per reaction | MNase | Profiles both open and closed genomic loci simultaneously, combined with ChIP specificity to enrich histone modification-associated DNA fragments. | Traditional method requires a large number of cells. | [65] |
| MH-seq | Paired-end sequencing | 10–20 million cells | MNase | Simple procedures, enables detection of distinct types of open chromatin. | Traditional method requires more cells, it is not easy in plants to establish Single-cell-based MH-seq, application in plants has limitations, high requirements for nuclear quality. | [66] |
| Array-seq | Paired-end sequencing | 10–20 million cells | MNase | Reveals linker length and array regularity in unmappable areas. | Traditional method requires more cells, titration test required. | [44] |
| MRE-seq | Paired-end sequencing | Need to test | Methylation-sensitive restriction enzymes | The methylation status of most repeats can be revealed, the methylation state of a local region or a single CPG can be addressed, MREs are inexpensive. | The recognition range of methylation events is limited, and only those within MRE recognition sites can be detected. | [67] |
| Database | Description | Data Type | Species | Source | Reference |
|---|---|---|---|---|---|
| GTRD (Gene Transcription Regulation Database) | The largest integrated resource of data on transcription regulation in eukaryotes, which contains uniformly annotated and processed NGS data, the results of the meta-analysis, and the sets of non-redundant and reproducible TFBSs for each TF. | ChIP-seq, ChIP-exo, DNase-seq, MNase-seq, ATAC-seq, RNA-seq | Homo sapiens, Mus musculus, Rattus norvegicus, Danio rerio, Caenorhabditis elegans, Drosophila melanogaster, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Arabidopsis thaliana | http://gtrd.biouml.org/ (accessed on 8 January 2021) | [81] |
| NPRD (Nucleosome Positioning Region Database) | It is compiling the available experimental data on locations and characteristics of nucleosome formation sites (NFSs), and is the first curated NFS-oriented database. | The type used in original paper | S. cerevisiae, Homo sapiens, Simian virus, Mus musculus, Drosophila melanogaster, Rattus norvegicus, Tetrahymena thermophila, Chironomus tentans, Mouse mammary tumor virus, Xenopus laevis (African clawed frog), Oryctolagus cuniculus (rabbit) | http://srs6.bionet.nsc.ru/srs6/ (accessed on 20 June 2022) | [86] |
| ChIP-Atlas | An integrative, comprehensive database to explore public Epigenetic dataset, covers almost all public data archived in Sequence Read Archive of NCBI, EBI, and DDBJ with over 224,000 experiments. | ChIP-Seq, DNase-Seq, ATAC-Seq, Bisulfite-Seq | H. sapiens, M. musculus, R. norvegicus, D. melanogaster, C. elegans, S. cerevisiae | https://chip-atlas.org/ (accessed on 4 January 2021) | [87] |
| CistromeDB | A resource of human and mouse cis-regulatory information, which map the genome-wide locations of transcription factor binding sites, histone post-translational modifications, and regions of chromatin accessible to endonuclease activity. | ChIP-seq, DNase-seq, ATAC-seq | H. sapiens, M. musculus | http://cistrome.org/db/#/ (accessed on 20 June 2021) | [88] |
| ENCODE | Integrative-level annotations integrate multiple types of experimental data and ground level annotations. Ground-level annotations are derived directly from the experimental data, typically produced by uniform processing pipelines. | ChIP-seq, DNase-seq, ATAC-seq, TFChIP-seq, RNA-seq, eCLIP-seq, ChIA-PET, Hi-C, RRBS, WGBS, RAMPAGE | H. sapiens, M. musculus, D. melanogaster, C. elegans | https://www.encodeproject.org (accessed on 30 January 2019) | [89] |
| ChIPBase | Decoding the encyclopedia of transcriptional regulations of ncRNAs and PCGs. | ChIP-seq, ChIP-exo, ChIP-nexus, MNChIP-seq | H. sapiens, M. musculus, R. norvegicus, D. rerio, X. tropicalis, C. elegans, D. melanogaster, S. cerevisiae, A. thaliana, G. gallus | https://rna.sysu.edu.cn/chipbase3/index.php (accessed on 1 July 2021) | [90] |
| ReMap 2020 3rd release | Information of regulatory regions from an integrative analysis of Human and Arabidopsis DNA-binding sequencing experiments. | ChIP-seq, ChIP-exo, ChIP-nexus, DAP-seq | H. sapiens, A. thaliana | http://remap.univ-amu.fr (accessed on 7 January 2022) | [91] |
| Factorbook | A transcription factor (TF)-centric repository of all ENCODE ChIP-seq datasets on TF-binding regions, as well as the rich analysis results of these data. | ChIP-seq | H. sapiens, M. musculus | http://factorbook.org (accessed on 20 June 2022) | [92] |
| NucMap | Genome-wide nucleosome positioning map across different species. | MNase-Seq | Arabidopsis thaliana, Caenorhabditis elegans, Candida albicans, Danio rerio, Drosophila melanogaster, Homo sapiens, Mus musculus, Neurospora crassa, Oryza sativa, Plasmodium falciparum, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Trypanosoma brucei, Xenopus laevis, Zea mays | http://bigd.big.ac.cn/nucmap (accessed on 4 April 2022) | [93] |
| NucPosDB | Database reporting experimental nucleosome maps in vivo across different cell types and conditions, cell-free DNA (cfDNA) datasets in people and model organisms, processed stable-nucleosome regions, as well as software for computational analysis and modeling of nucleosome positioning and “nucleosomics” analysis for medical diagnostics. | MNase-seq, ChIP-seq, MH-seq, MPE-seq, MiSeq, NOME-seq, RED-seq, Nanopore-seq, Fiber-seq, Ucleosome-scale mapping of 3D genome contact, Micro-C | S. cerevisiae, M. musculus, H. sapiens, D. melanogaster, A. thaliana, S. pombe, C. elegans, N. crassa | https://generegulation.org/nucposdb/ (accessed on 20 June 2022) | [94] |
| Algorithm | Web Server/GUI | Feature | Input Dataset | Languages | Source | Reference |
|---|---|---|---|---|---|---|
| CAESAR | +/− | Connecting epigenomics and chromatin organization at the nucleosome resolution. | Epigenomic features and Hi-C contact maps | Python | https://github.com/liu-bioinfo-lab/caesar (accessed on 1 March 2022) | [82] |
| Factor-agnostic chromatin occupancy profiles from MNase | +/− | Links changes in chromatin at nucleotide resolution with transcriptional regulation. | MNase-seq and RNA-seq data | Python, Shell | https://github.com/HarteminkLab/cadmium-paper (accessed on 18 April 2021) | [85] |
| NucHMM | +/− | Identifies functional nucleosome states associated with cell type-specific combinatorial histone marks and nucleosome organization features. | MNase-seq and ChIP-seq data | Python, C++, Makefile | https://github.com/KunFang93/NucHMM (accessed on 2 June 2022) | [83] |
| ProbC | +/− | Decomposes Hi-C and Micro-C interactions by known chromatin marks at genome and chromosome levels. | Hi-C and Micro-C data | Python | http://www.github.com/seferlab/probc (accessed on 19 March 2022) | [84] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, S.; Lu, Y.; Tan, S.; Li, R.; Gao, Y.; Li, K.; Zhang, Y. Nucleosome-Omics: A Perspective on the Epigenetic Code and 3D Genome Landscape. Genes 2022, 13, 1114. https://doi.org/10.3390/genes13071114
Kong S, Lu Y, Tan S, Li R, Gao Y, Li K, Zhang Y. Nucleosome-Omics: A Perspective on the Epigenetic Code and 3D Genome Landscape. Genes. 2022; 13(7):1114. https://doi.org/10.3390/genes13071114
Chicago/Turabian StyleKong, Siyuan, Yuhui Lu, Shuhao Tan, Rongrong Li, Yan Gao, Kui Li, and Yubo Zhang. 2022. "Nucleosome-Omics: A Perspective on the Epigenetic Code and 3D Genome Landscape" Genes 13, no. 7: 1114. https://doi.org/10.3390/genes13071114
APA StyleKong, S., Lu, Y., Tan, S., Li, R., Gao, Y., Li, K., & Zhang, Y. (2022). Nucleosome-Omics: A Perspective on the Epigenetic Code and 3D Genome Landscape. Genes, 13(7), 1114. https://doi.org/10.3390/genes13071114