
Identification of miRNAs in Response to Sweet Potato Weevil (Cylas formicarius) Infection by sRNA Sequencing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Infection Treatment and Sample Collection
2.3. Library Preparation and Small RNA Sequencing
2.4. Identification of miRNA
2.5. Identification of Differentially Expressed miRNA
2.6. Prediction of miRNA Targets
2.7. qRT-PCR Analysis
3. Results
3.1. High-Throughput Sequencing Data Analysis
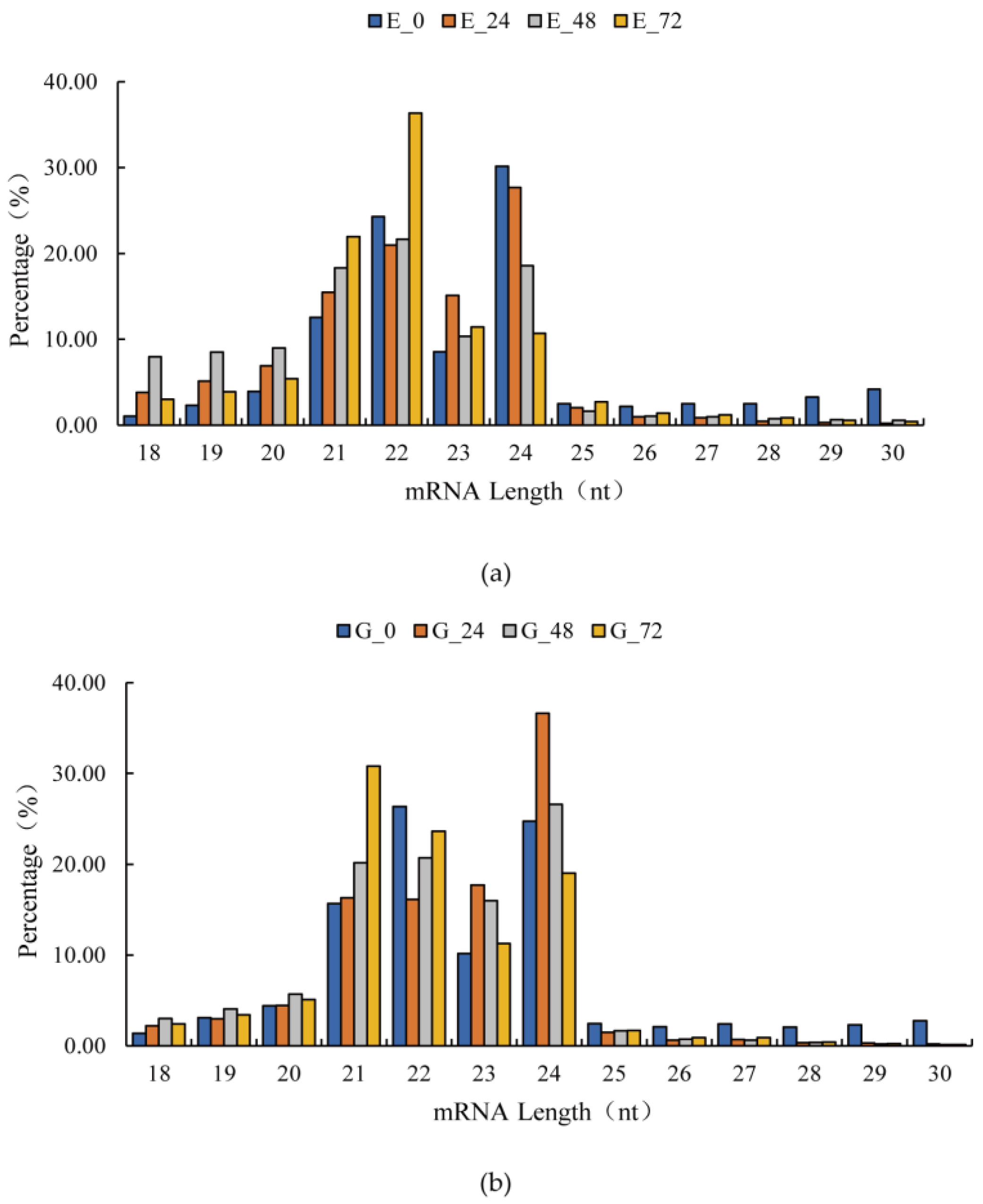
3.2. Identification of Known miRNAs and Novel miRNAs
3.3. Analysis of Differentially Expressed miRNAs
3.4. Predicted miRNA Target Genes and GO/KEGG Enrichment Analyses
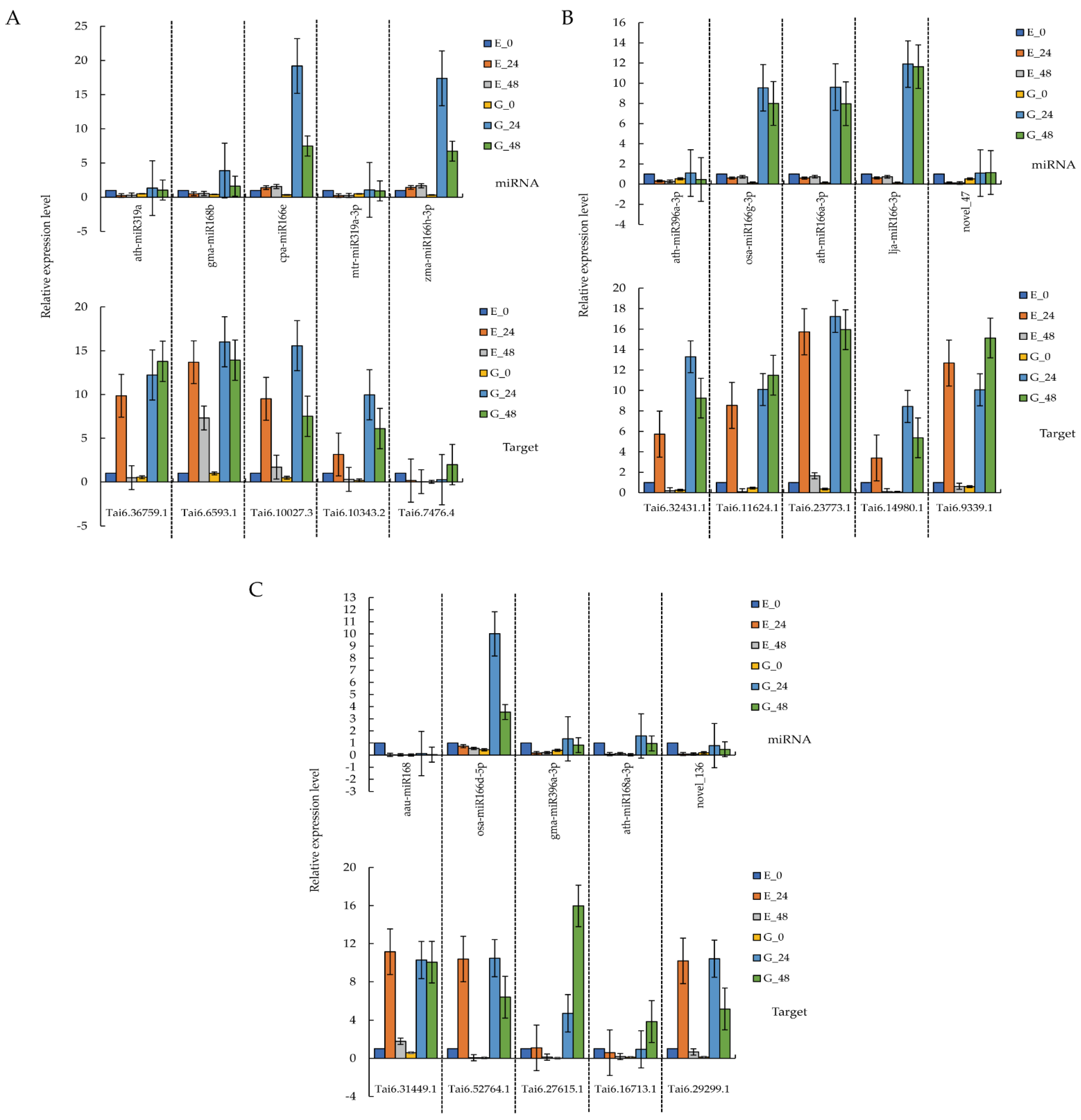
3.5. qRT-PCR Verification
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, P.J.; Song, Q.Q.; Tan, Q.L.; Cheng, Q.; Li, J.H.; Pang, X.H.; Zhou, Q.G.; Lü, P.; Ou, K.W.; Lu, Y.F.; et al. Bioinformatics analysis of microRNAs and prediction of target genes associated with cold tolerance in sugarcane. Guihaia 2021, 1–17. Available online: http://kns.cnki.net/kcms/detail/45.1134.Q.20210715.1618.002.html (accessed on 28 May 2022).
- Liu, Y.; Zhang, X.Y.; Xu, M.Y.; Zheng, H.Y.; Zou, J.J.; Zhang, L.; Wang, L. Global small RNA transcriptome profiling of rice under drought stress. J. Agric. Sci. Technol. 2021, 23, 23–32. [Google Scholar]
- Pandey, P.; Srivastava, P.K.; Pandey, S.P. Prediction of plant miRNA targets. Methods Mol. Biol. 2019, 1932, 99–107. [Google Scholar]
- Liu, J.; Liu, X.N.; Zhang, S.J.; Liang, S.S.; Luan, W.J.; Ma, X. TarDB: An online database for plant miRNA targets and miRNA-triggered phased siRNAs. BMC Genom. 2021, 22, 348. [Google Scholar] [CrossRef]
- Anushree, N.; Shivaprasad, P.V. Regulation of plant miRNA biogenesis. Proc. Indian Natl. Sci. Acad. 2017, 95, 439–453. [Google Scholar] [CrossRef]
- Narjala, A.; Nair, A.; Tirumalai, V.; Hari Sundar, G.V.; Shivaprasad, P.V. A conserved sequence signature is essential for robust plant miRNA biogenesis. Nucleic Acids Res. 2020, 48, 3103–3118. [Google Scholar] [CrossRef]
- Zhang, Y.-C.; Yu, Y.; Wang, C.-Y.; Li, Z.-Y.; Liu, Q.; Xu, J.; Liao, J.-Y.; Wang, X.-J.; Qu, L.-H.; Chen, F.; et al. Overexpression of microRNA OsmiR397 improves rice yield by increasing grain size and promoting panicle branching. Nat. Biotechnol. 2013, 31, 848. [Google Scholar] [CrossRef]
- Yu, N.; Niu, Q.W.; Ng, K.H.; Chua, N.H. The role of miR156/SPLs modules in Arabidopsis lateral root development. Plant J. Cell Mol. Biol. 2015, 83, 673–685. [Google Scholar] [CrossRef]
- Jiang, J.X.; Zhang, J.Y.; Li, Y.L.; Jiang, M.Y.; Zhu, J.F.; Zhou, X.R.; Wang, W.R.; Sun, C.C.; Yang, L.Y. Research progress of plant microRNAs in stress response. Mol. Plant Breed 2021, 1–14. Available online: http://kns.cnki.net/kcms/detail/46.1068.S.20210806.1107.005.html (accessed on 28 May 2022).
- Tong, B.; Zhan, J.; Wang, A.Q.; Xiao, D. HeLF: Research progress of miRNA in the regulation of programmed cell death in plants. Mol. Plant Breed 2022, 1–26. Available online: http://kns.cnki.net/kcms/detail/46.1068.S.20220104.1521.007.html (accessed on 28 May 2022).
- Lv, X.M.; Zhang, W.Y.; Zhang, H.H.; Liang, Z.S.; Chen, H.M. Advances of miRNA mediate regulatory roles in plant-microbe interaction. Chin. J. Biotech. 2022, 38, 1695–1705. [Google Scholar] [CrossRef]
- Hao, D.C.; Yang, L.; Xiao, P.G.; Liu, M. Identification of Taxus microRNAs and their targets with high-throughput sequencing and degradome analysis. Physiol. Plant. 2012, 146, 388–403. [Google Scholar] [CrossRef]
- Zheng, X.Y.; Chen, M.; Li, H.; Zhao, S.J. Research methods of miRNA in plants and research progress of miRNA in medicinal plants. Acta Pharm. Sin. 2021, 56, 3460–3472. [Google Scholar]
- Yang, C.H.; Li, D.Y.; Mao, D.H.; Liu, X.; Ji, C.J.; Li, X.B.; Zhao, X.F.; Cheng, Z.K.; Chen, C.Y.; Zhu, L.H. Overexpression of microRNA319 impacts leaf morphogenesis and leads to enhanced cold tolerance in rice (Oryza sativa L.). Plant Cell Environ. 2013, 36, 2207–2218. [Google Scholar] [CrossRef]
- Wamiq, G.; Khan, J.A. Overexpression of ghr-miR166b generates resistance against Bemisia tabaci infestation in Gossypium hirsutum plants. Planta 2018, 247, 1175–1189. [Google Scholar] [CrossRef]
- Li, M.; Yu, B. Recent advances in the regulation of plant miRNA biogenesis. RNA Biol. 2021, 18, 2087–2096. [Google Scholar] [CrossRef]
- Rohit, D.; Ananya, M.; Shrabani, B.; Pallob, K. Plant miRNA responses under temperature stress. Plant Gene 2021, 28, 100317. [Google Scholar]
- Mao, Y.B.; Chen, D.Y.; Chen, X.Y. Advances in research of plant non-coding RNAs and RNAi-based technology against insects herbivore. Bull. Chin. Acad. Sci. 2017, 32, 814–821. [Google Scholar]
- Pandey Shree, P.; Shahi, P.; Gase, K.; Baldwin, I.T. Herbivory-induced changes in the small-RNA transcriptome and phytohormone signaling in Nicotiana attenuata. Proc. Natl. Acad. Sci. USA 2008, 105, 4559–4564. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Wang, L.J.; Su, W.J.; Chai, S.S.; Yang, X.S. Research development of the control of sweetpotato weevil. Hubei Agric. Sci. 2018, 57, 9–12. [Google Scholar]
- Zhang, S.W.; Talekar, N.S.; Li, Z.Y.; Sun, Y.X. Selection behaviors of Cylas formicarius (F.) adult to different parts of sweetpotato plant. J. Yunnan Univ. Nat. Sci. Ed. 2008, 30, 127–129. [Google Scholar]
- Chen, F.R.; Yang, X.J.; Zhang, L.S.; Lu, T.; Yang, J.Y.; Lin, W.D. Application of sex pheromone in controlling sweetpotato weevil. Fujian J. Agric. Sci. 2001, 1, 16–19. [Google Scholar]
- Xu, J.; Su, Z.P.; Su, H.R.; Zhu, H.B.; Cheng, S.Y.; Wu, Z.W. Insecticide controlling sweet poato weevil: Selection and field efficacy. Chin. Agric. Sci. Bull. 2021, 37, 153–158. [Google Scholar]
- Chen, F.R.; Yang, X.J.; Zhang, L.S.; Lu, T.; Lin, G.F.; Yang, J.Y.; Huang, X.H. A Study and application of the integrated control technologic system for the control of sweetpotato weevil. Acta Agric. Univ. Jiangxiensis Nat. Sci. Ed. 2002, 4, 445–447. [Google Scholar]
- Hua, J.F.; Fu, Y.J.; Zhou, Q.L.; Huang, Y.M.; Li, H.F.; Chen, T.Y.; Ma, D.F.; Li, Z.Y. Three chemosensory proteins from the sweet potato weevil, Cylas formicarius, are involved in the perception of host plant volatiles. Pest Manag. Sci. 2021, 77, 4497–4509. [Google Scholar] [CrossRef]
- Yu, H.B.; Shen, J.W.; Ma, J.; Ma, H.J.; Chen, S.L. The study on rDNA ITS-1 variation of Cylas formicarius (Coleoptera: Brentidae) populations and its invasive sources in China. Chin. Agric. Sci. Bull. 2011, 27, 282–287. [Google Scholar]
- Lei, J.; Zhang, M.; Zhang, J.Z.; Wang, L.J.; Chai, S.S.; Jin, X.J.; Cheng, X.L.; Yang, Y.Y.; Yang, X.S. Chemotaxis test of sweetpotato weevil (Cylas formicarius) to sweet potato varieties. Chin. Agric. Sci. Bull. 2021, 37, 20–24. [Google Scholar]
- Rao, S.F.; Liu, X.; Liao, Y.Y.; Wang, Y.R.; Zhu, H.B.; Yang, Z.Y.; Hu, B.; Hou, X.L. Transcriptomic analysis of the sweetpotato ’Guangshu 87’ under the feeding treatment by weevil. Plant Physiol. 2020, 56, 1484–1492. [Google Scholar]
- Yang, Y.H.; Liu, Q.C.; Zhai, H. Research progress in sweetpotato miRNA. J. Jiangsu North Univ. Agric. Sci. 2021, 39, 42–45. [Google Scholar]
- Sun, R.; Guo, T.; Cobb, J.; Wang, Q.; Zhang, B. Role of microRNAs during flower and storage root development in sweet potato. Plant Mol. Biol. Rep. 2015, 33, 1731–1739. [Google Scholar] [CrossRef]
- Kuo, Y.W.; Lin, J.S.; Li, Y.C.; Jhu, M.Y.; King, Y.C.; Jeng, S.T. MicroR408 regulates defense response upon wounding in sweet potato. J. Exp. Bot. 2019, 70, 469–483. [Google Scholar] [CrossRef]
- Ben, L. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinform. 2010, 32, 11–17. [Google Scholar]
- Kozomara, A.; Sam, G.-J. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Wen, M.; Shen, Y.; Shi, S.H.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinf. 2012, 13, 140. [Google Scholar] [CrossRef] [Green Version]
- Friedländer Marc, R.; Mackowiak Sebastian, D.; Li, N.; Chen, W.; Nikolaus, R. MiRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Love Michael, I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Fahlgren, N.; Carrington James, C. MiRNA target prediction in plants. Methods Mol. Biol. 2010, 592, 51–57. [Google Scholar]
- Young Matthew, D.; Wakefield Matthew, J.; Smyth Gordon, K.; Alicia, O. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.Z.; Cai, T.; Olyarchuk John, G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Zhu, T.Y.; Xu, K.S.; Zhang, G.; Li, Z.; Xue, C. Optical calibration and result analysis of real-time fluorescence quantitative PCR instrument. Metrol. Meas. Tech. 2019, 46, 39–42. [Google Scholar]
- Tang, C.; Han, R.P.; Zhou, Z.K.; Yang, Y.Y.; Zhu, M.K.; Xu, T.; Wang, A.M.; Li, Z.Y.; Dong, T.T. Identification of candidate miRNAs related in storage root development of sweet potato by high throughput sequencing. J. Plant Physiol. 2020, 251, 153224. [Google Scholar] [CrossRef]
- Zhao, M.L.; Tian, M.Y.; Li, L.; Ren, Y.J. Present situation and prospect of microRNA involved in the regulation of plant stress resistance. Genom. Appl. Biol. 2020, 39, 5255–5262. [Google Scholar]
- Mu, H.F.; Qi, W.; Li, Y.; Wu, Y. Research advances of microRNAs in plant resistance to pathogens. Mod. Agric. Sci. Technol. 2016, 23, 144–147. [Google Scholar]
- Li, C.D.; Wong Annette, Y.P.; Wang, S.; Jia, Q.; Chuang, W.P.; Bendena William, G.; Tobe Stephen, S.; Yang Seung, H.; Chung, G.; Chan, T.F.; et al. miRNA-mediated interactions in and between plants and insects. Int. J. Mol. Sci. 2018, 19, 3239. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.Y. Identication of Chilling-Responsive microRNAs and Their Targets in Sweet Potato (Ipomoea Batatas Lam.) during the Storage; Jiangsu Normal University: Xuzhou, China, 2017. [Google Scholar]
- Shi, X.W. Identification and Analysis of microRNA Related to Adversity Stress and Anthocyanin Biosynthesis in Sweet Potato; Shanxi Agricultural University: Jinzhong, China, 2018. [Google Scholar]
- Liu, X.Y. Establishment of CRISPR/Cas9 System and Study Onregulation Mechanism of miR2111 in Anthocyanin Accumulation of Sweet Potato; Shanxi Agricultural University: Jinzhong, China, 2020. [Google Scholar]
- Han, J.T.; Feng, G.Y.; Shuai, Y.; Jiao, Y.J.; Zhang, X.Q. Advances in research of miRNA156 and targeted SPL gene in plants. Pratac. Sci. 2021, 38, 890–902. [Google Scholar]
- Ma, Z.X.; Hu, X.P.; Cai, W.J.; Huang, W.H.; Zhou, X.; Luo, Q.; Yang, H.Q.; Wang, J.W.; Huang, J. Arabidopsis miR171-targeted scarecrow-like proteins bind to GT cis-elements and mediate gibberellin-regulated chlorophyll biosynthesis under light conditions. PLoS Genet. 2014, 10, e1004519. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.Y.; Zhao, B.; Chao, L.M.; Chen, D.Y.; Cui, W.R.; Mao, Y.B.; Wang, L.J.; Chen, X.Y. Interaction between two timing microRNAs controls trichome distribution in Arabidopsis. PLoS Genet. 2014, 10, e1004266. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.F. miR156-Mediated Regulatory Mechanism of BPH Resistance in Rice; Zhejiang A&F University: Lin’an City, China, 2017. [Google Scholar]
- Li, Y.H.; Lin, Z.Q.; Zhu, T.F.; Huang, Z.M.; Guo, X.; Su, J. Expression pattern of insect resistance related gene PhSPL17 in phyllostachys heterocycle during on-and off-year. J. Fujian Agric. For. Univ. Nat. Sci. 2019, 48, 597–604. [Google Scholar]
- Yu, N.; Cai, W.J.; Wang, S.C.; Shan, C.M.; Wang, L.J.; Chen, X.Y. Temporal control of trichome distribution by microRNA156-targeted SPL genes in Arabidopsis thaliana. Plant Cell 2010, 22, 2322–2335. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.Y. Physiological Response of Rice to Brown Planthopper and the Study of Small Brown Planthopper microRNA; Zhejiang University: Hangzhou, China, 2014. [Google Scholar]
- Li, J.Y. Molecular Mechanism of Cotton in Response to Whitefly Ifestation and Epigenomics Basis of Cotton Somatic Embryogenesis. J. Huazhong Agric. Univ. 2019. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library | Treatment | Sample ID | Total Reads | Clean Reads | Percentage (%) | Mapped Genome | Percentage (%) |
|---|---|---|---|---|---|---|---|
| Eshu 6 | E_0 | E_0_1 | 13,505,817 | 8,874,325 | 65.71 | 7,046,454 | 66.39 |
| E_0_2 | 13,547,525 | 10,174,405 | 75.10 | 4,838,795 | 50.56 | ||
| E_0_3 | 16,468,157 | 13,888,078 | 84.33 | 6,237,540 | 46.92 | ||
| Mean | 14,507,166 | 10,978,936 | 75.68 | 6,040,930 | 54.62 | ||
| E_24 | E_24_1 | 16,990,361 | 15,477,110 | 91.09 | 5,980,088 | 45.94 | |
| E_24_2 | 12,898,360 | 11,156,704 | 86.50 | 8,128,053 | 69.23 | ||
| E_24_3 | 14,544,136 | 12,618,934 | 86.76 | 7,616,186 | 66.19 | ||
| Mean | 14,810,952 | 13,084,249.33 | 88.34 | 7,241,442 | 60.45 | ||
| E_48 | E_48_1 | 16,179,390 | 13,552,999 | 83.77 | 9,147,300 | 68.44 | |
| E_48_2 | 12,901,334 | 9,569,488 | 74.17 | 10,439,875 | 57.02 | ||
| E_48_3 | 12,022,565 | 4,651,802 | 38.69 | 12,267,059 | 71.56 | ||
| Mean | 13,701,096 | 9,258,096.333 | 67.57 | 10,618,078 | 65.67 | ||
| E_72 | E_72_1 | 15,711,300 | 13,017,803 | 82.86 | 2,910,452 | 62.57 | |
| E_72_2 | 14,969,018 | 13,776,032 | 92.03 | 5,994,652 | 67.55 | ||
| E_72_3 | 16,047,099 | 13,294,682 | 82.85 | 5,389,906 | 53.80 | ||
| Mean | 15,575,806 | 13,362,839 | 85.79 | 4,765,003 | 61.31 | ||
| Guang 87 | G_0 | G_0_1 | 15,031,654 | 13,592,390 | 90.43 | 6,773,030 | 56.00 |
| G_0_2 | 13,460,140 | 10,613,259 | 78.85 | 6,516,875 | 64.05 | ||
| G_0_3 | 13,994,674 | 11,699,455 | 83.60 | 7,906,581 | 67.58 | ||
| Mean | 14,162,156 | 11,968,368 | 84.51 | 7,065,495 | 62.54 | ||
| G_24 | G_24_1 | 17,559,872 | 17,142,807 | 97.62 | 6,482,848 | 58.11 | |
| G_24_2 | 14,141,613 | 12,140,937 | 85.85 | 6,207,694 | 45.06 | ||
| G_24_3 | 13,623,060 | 11,507,160 | 84.47 | 7,465,376 | 63.02 | ||
| Mean | 15,108,181.67 | 13,596,968 | 90.00 | 6,718,639 | 55.40 | ||
| G_48 | G_48_1 | 13,554,175 | 11,845,193 | 87.39 | 7,608,730 | 60.30 | |
| G_48_2 | 14,180,870 | 13,364,466 | 94.24 | 9,247,207 | 68.23 | ||
| G_48_3 | 13,115,375 | 10,018,069 | 76.38 | 8,947,533 | 64.43 | ||
| Mean | 13,616,807 | 11,742,576 | 86.24 | 8,601,157 | 64.32 | ||
| G_72 | G_72_1 | 12,481,043 | 11,739,937 | 94.06 | 8,511,456 | 62.62 | |
| G_72_2 | 13,666,809 | 12,095,700 | 88.50 | 9,168,804 | 59.24 | ||
| G_72_3 | 19,897,861 | 18,307,922 | 92.01 | 8,574,439 | 70.62 | ||
| Mean | 15,348,571 | 14,047,853 | 91.53 | 8,751,566 | 64.16 |
| Mapped Mature Known miRNAs | Mapped Hairpin Known miRNAs | Mapped Mature Novel miRNAs | Mapped Hairpin Novel miRNAs | |
|---|---|---|---|---|
| Total | 407 | 908 | 298 | 307 |
| E_0_1 | 146 | 475 | 219 | 260 |
| E_0_2 | 161 | 482 | 237 | 265 |
| E_0_3 | 161 | 499 | 243 | 276 |
| E_24_1 | 228 | 577 | 268 | 293 |
| E_24_2 | 205 | 520 | 263 | 288 |
| E_24_3 | 197 | 516 | 266 | 288 |
| E_48_1 | 201 | 510 | 249 | 276 |
| E_48_2 | 211 | 545 | 244 | 277 |
| E_48_3 | 201 | 543 | 207 | 258 |
| E_72_1 | 192 | 490 | 226 | 268 |
| E_72_2 | 202 | 533 | 246 | 277 |
| E_72_3 | 152 | 385 | 205 | 252 |
| G_0_1 | 178 | 500 | 248 | 272 |
| G_0_2 | 153 | 491 | 243 | 269 |
| G_0_3 | 178 | 491 | 245 | 274 |
| G_24_1 | 177 | 514 | 273 | 293 |
| G_24_2 | 239 | 569 | 275 | 294 |
| G_24_3 | 229 | 587 | 274 | 297 |
| G_48_1 | 221 | 566 | 278 | 289 |
| G_48_2 | 221 | 570 | 271 | 290 |
| G_48_3 | 151 | 405 | 214 | 250 |
| G_72_1 | 208 | 578 | 265 | 292 |
| G_72_2 | 192 | 503 | 233 | 267 |
| G_72_3 | 246 | 585 | 269 | 301 |
| Samples Comparison | Differentially Expressed | Up-Regulated | Down-Regulated |
|---|---|---|---|
| E_0 vs. E_24 | 270 | 164 | 106 |
| E_0 vs. E_48 | 223 | 139 | 84 |
| E_0 vs. E_72 | 236 | 120 | 116 |
| G_0 vs. G_24 | 173 | 102 | 71 |
| G_0 vs. G_48 | 186 | 115 | 71 |
| G_0 vs. G_72 | 223 | 131 | 92 |
| E_24 vs. G_24 | 131 | 84 | 47 |
| E_48 vs. G_48 | 65 | 28 | 37 |
| E_72 vs. G_72 | 79 | 48 | 31 |
| miRNA Family | miRNA | Target_mRNA | Nr |
|---|---|---|---|
| MIR167_1 | bna-miR167d | Tai6.4195.1 | PREDICTED: glutamate–cysteine ligase, chloroplastic-like [Ipomoea nil] |
| bna-miR167d | Tai6.35623.1 | PREDICTED: glutamate–cysteine ligase, chloroplastic-like [Ipomoea nil] | |
| vvi-miR167c | Tai6.4195.1 | PREDICTED: glutamate–cysteine ligase, chloroplastic-like [Ipomoea nil] | |
| vvi-miR167c | Tai6.35623.1 | PREDICTED: glutamate–cysteine ligase, chloroplastic-like [Ipomoea nil] | |
| ptc-miR167f-5p | Tai6.4195.1 | PREDICTED: glutamate–cysteine ligase, chloroplastic-like [Ipomoea nil] | |
| ptc-miR167f-5p | Tai6.35623.1 | PREDICTED: glutamate–cysteine ligase, chloroplastic-like [Ipomoea nil] | |
| MIR156 | hbr-miR156 | Tai6.44728.1 | PREDICTED: protein SENESCENCE-ASSOCIATED GENE 21, mitochondrial-like [Ipomoea nil] |
| n_MIR318 | novel_318 | Tai6.52197.1 | PREDICTED: ethylene receptor 1 isoform X1 [Ipomoea nil] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, J.; Mei, Y.; Jin, X.; Liu, Y.; Wang, L.; Chai, S.; Cheng, X.; Yang, X. Identification of miRNAs in Response to Sweet Potato Weevil (Cylas formicarius) Infection by sRNA Sequencing. Genes 2022, 13, 981. https://doi.org/10.3390/genes13060981
Lei J, Mei Y, Jin X, Liu Y, Wang L, Chai S, Cheng X, Yang X. Identification of miRNAs in Response to Sweet Potato Weevil (Cylas formicarius) Infection by sRNA Sequencing. Genes. 2022; 13(6):981. https://doi.org/10.3390/genes13060981
Chicago/Turabian StyleLei, Jian, Yuqin Mei, Xiaojie Jin, Yi Liu, Lianjun Wang, Shasha Chai, Xianliang Cheng, and Xinsun Yang. 2022. "Identification of miRNAs in Response to Sweet Potato Weevil (Cylas formicarius) Infection by sRNA Sequencing" Genes 13, no. 6: 981. https://doi.org/10.3390/genes13060981
APA StyleLei, J., Mei, Y., Jin, X., Liu, Y., Wang, L., Chai, S., Cheng, X., & Yang, X. (2022). Identification of miRNAs in Response to Sweet Potato Weevil (Cylas formicarius) Infection by sRNA Sequencing. Genes, 13(6), 981. https://doi.org/10.3390/genes13060981