
scHiCEmbed: Bin-Specific Embeddings of Single-Cell Hi-C Data Using Graph Auto-Encoders


Abstract
:1. Introduction
2. Materials and Methods
2.1. Hi-C Data Processing
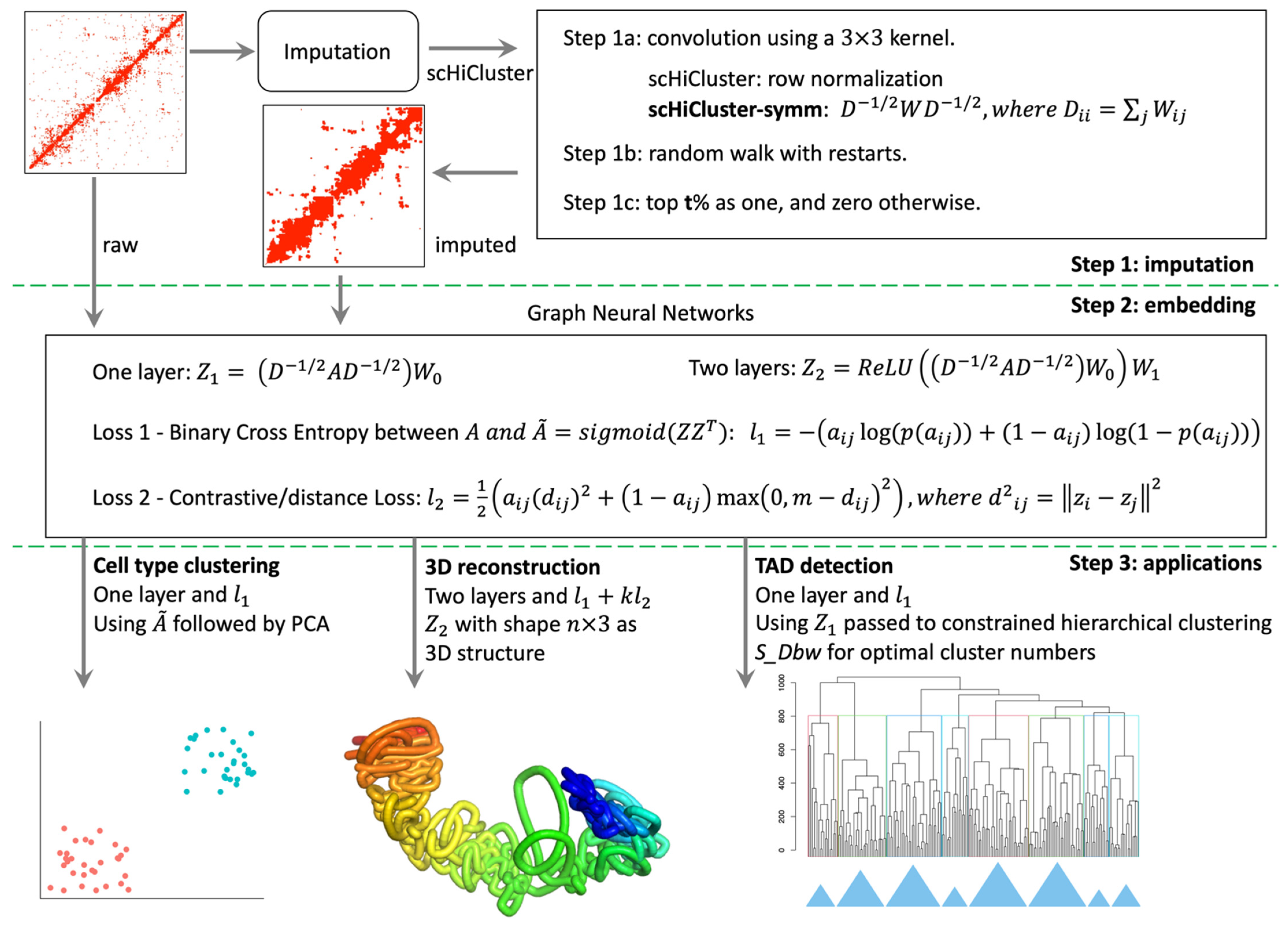
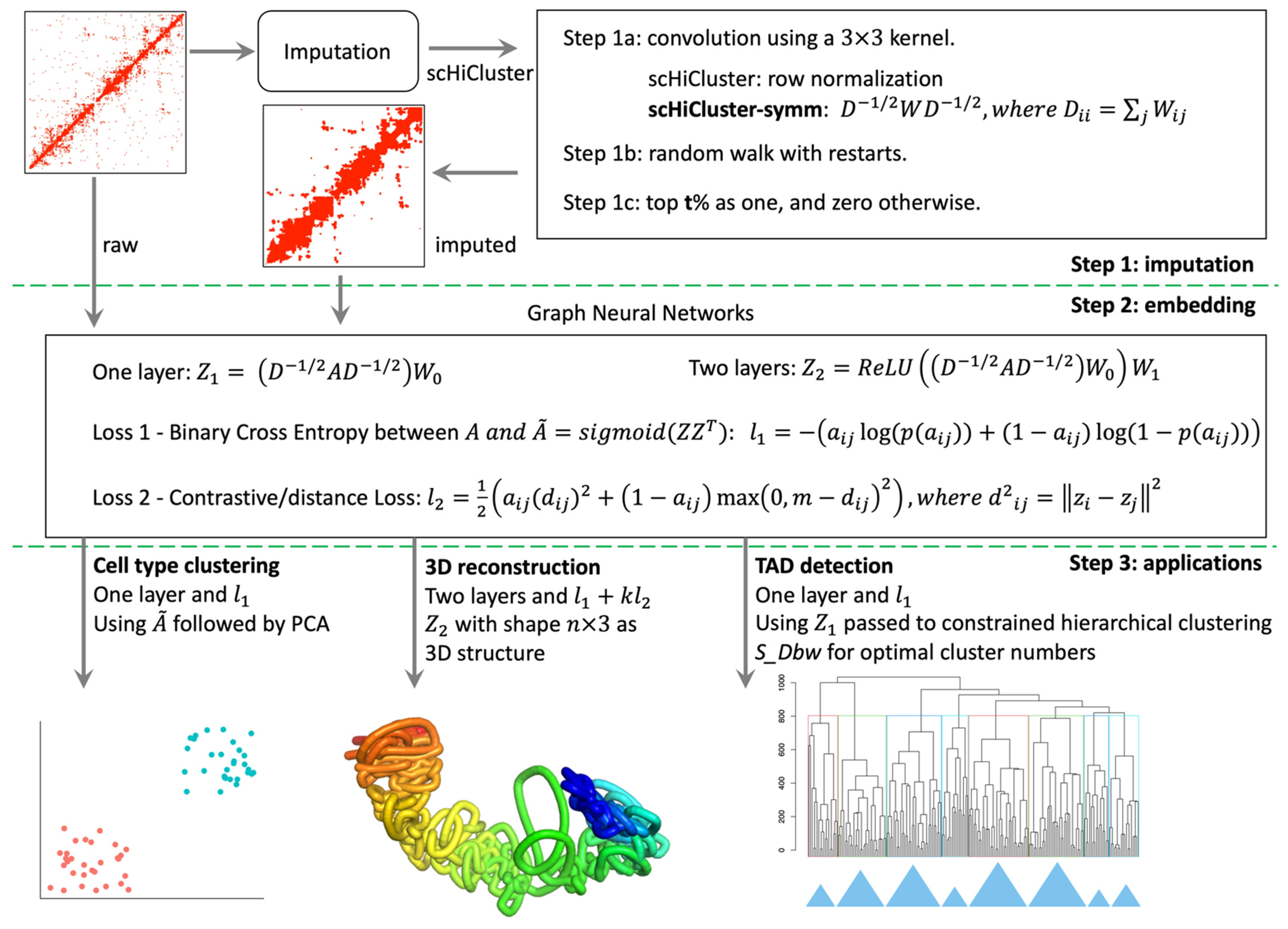
2.2. Overview of the scHiCEmbed Pipeline
2.3. Imputation of Single-Cell Hi-C Contact Matrices
2.4. Bin-Specific Embeddings Using Graph Auto-Encoders
2.5. Training, Validation, and Blind Test
2.6. Tuning Hyperparameters
2.7. Cell Type Clustering
2.8. 3D Genome Reconstruction
2.9. Calling TADs Based on Embeddings
2.10. TADs and Methylation
3. Results
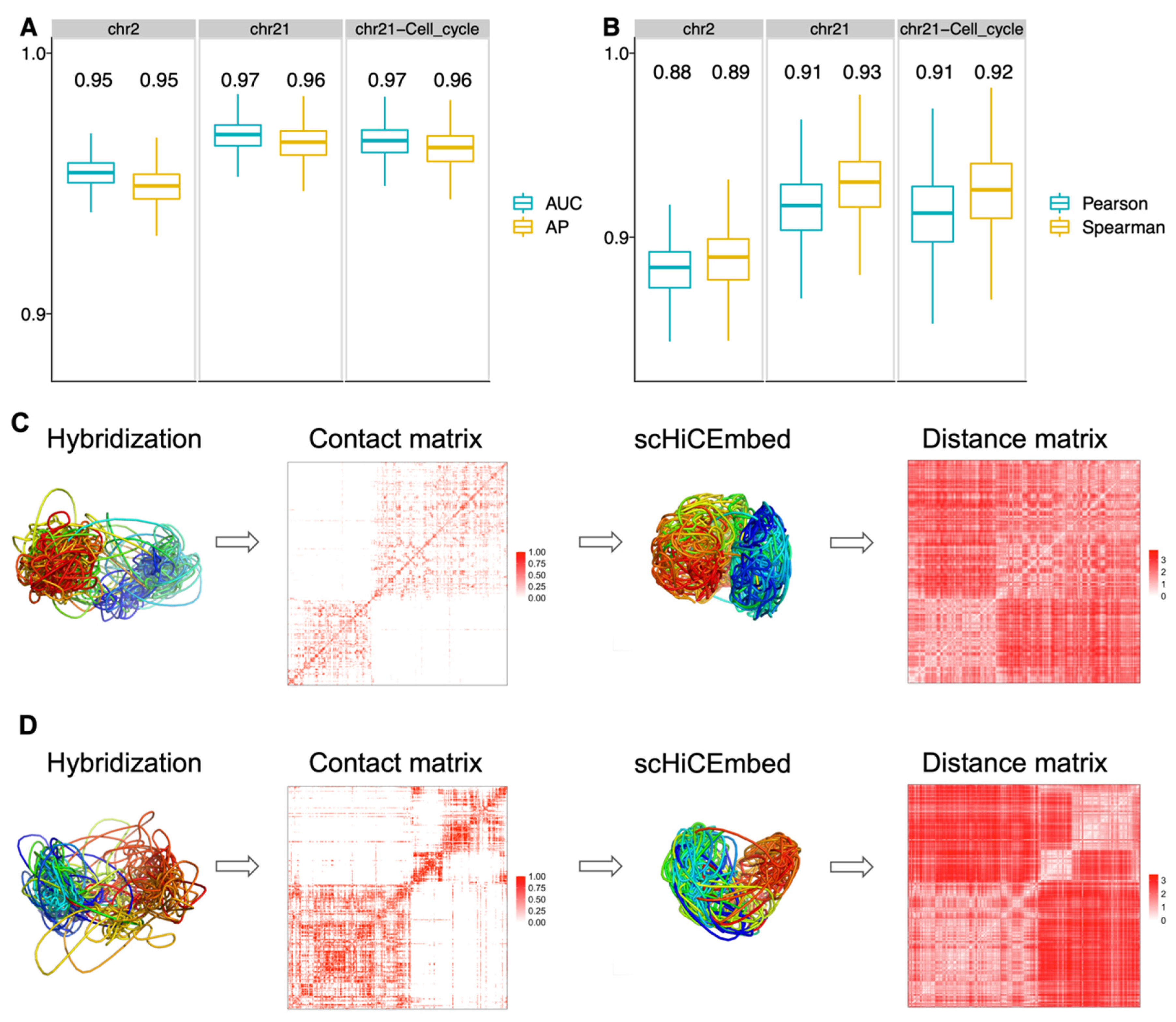
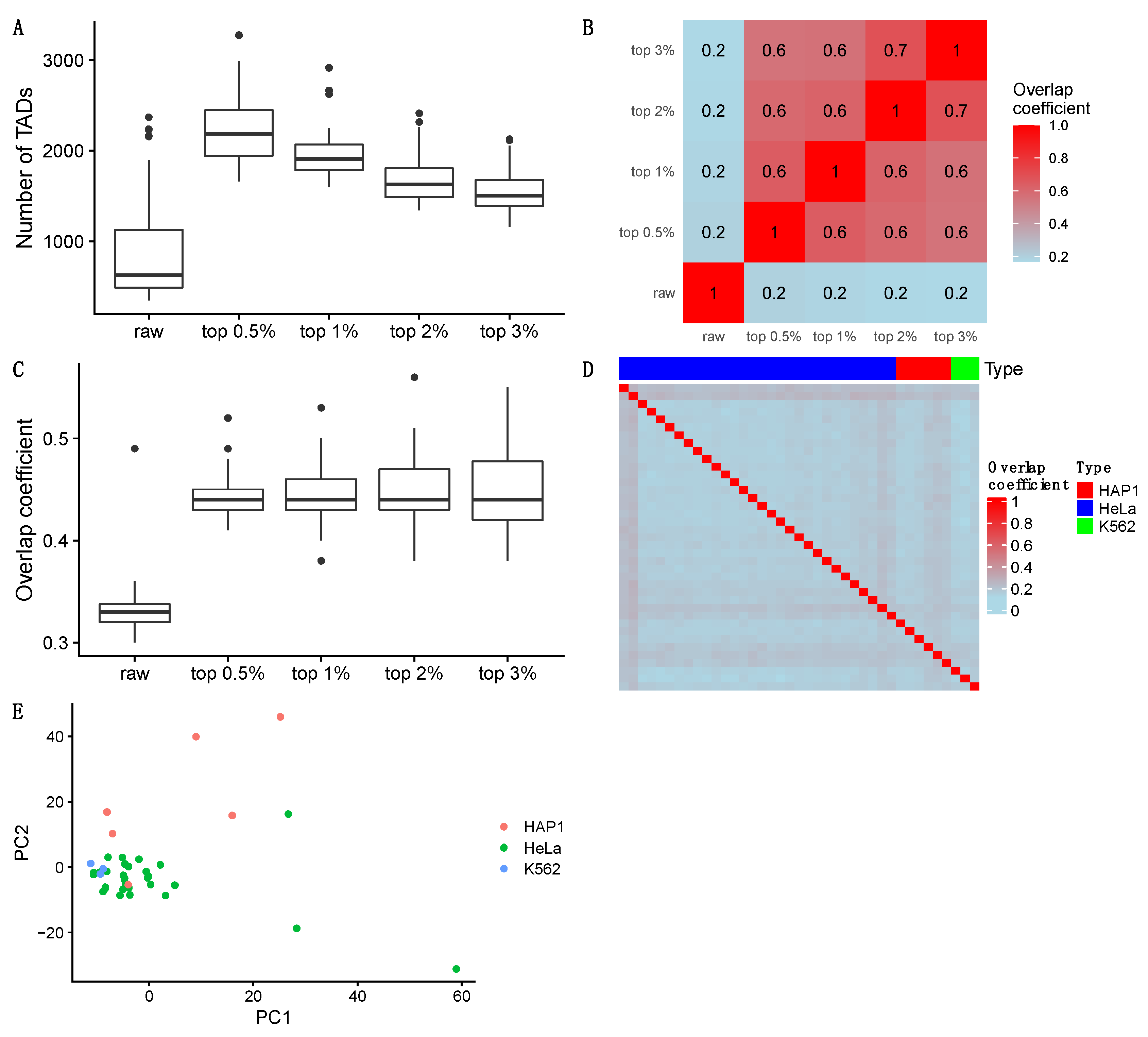
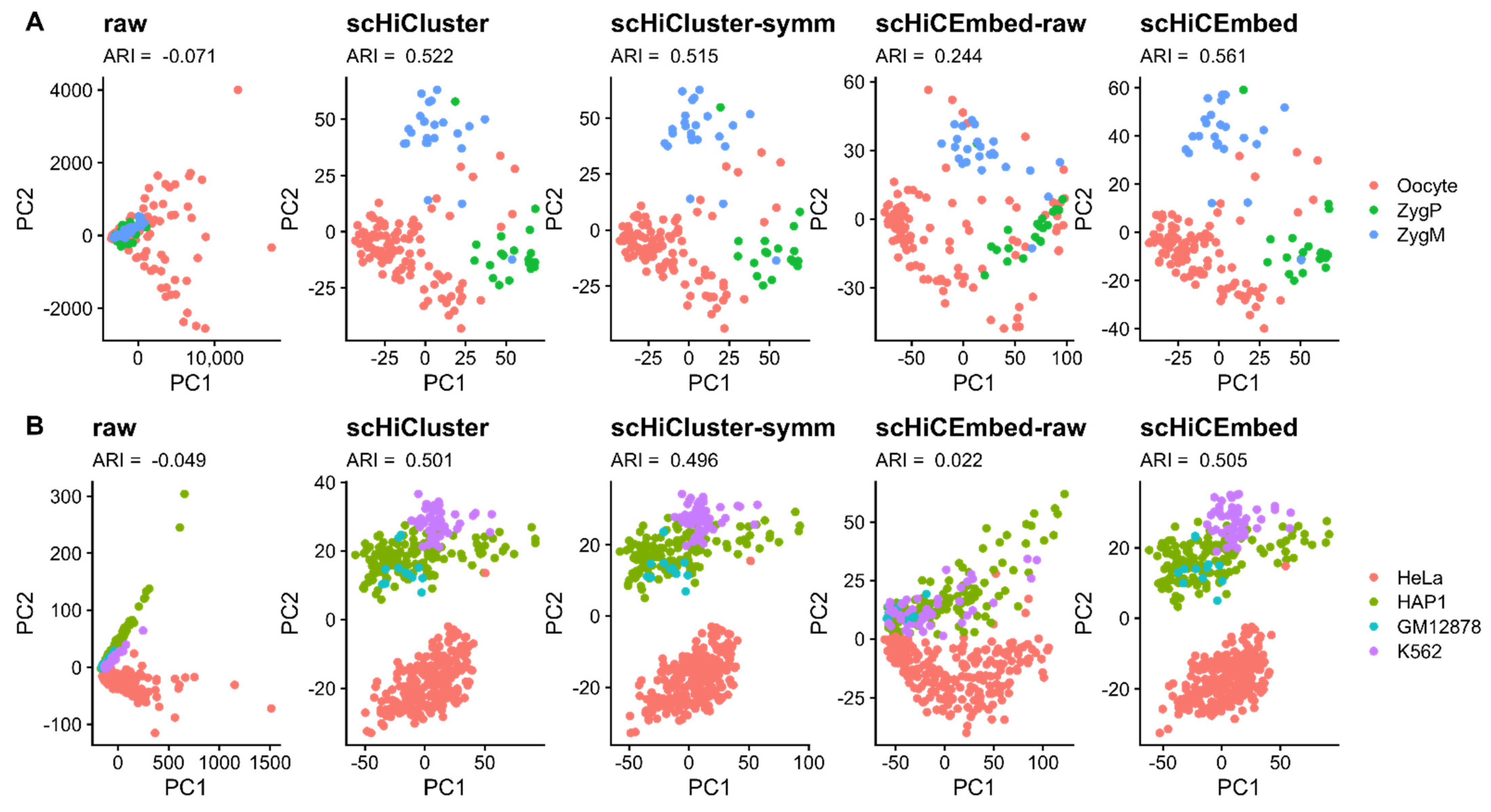
3.1. Cell Type Clustering
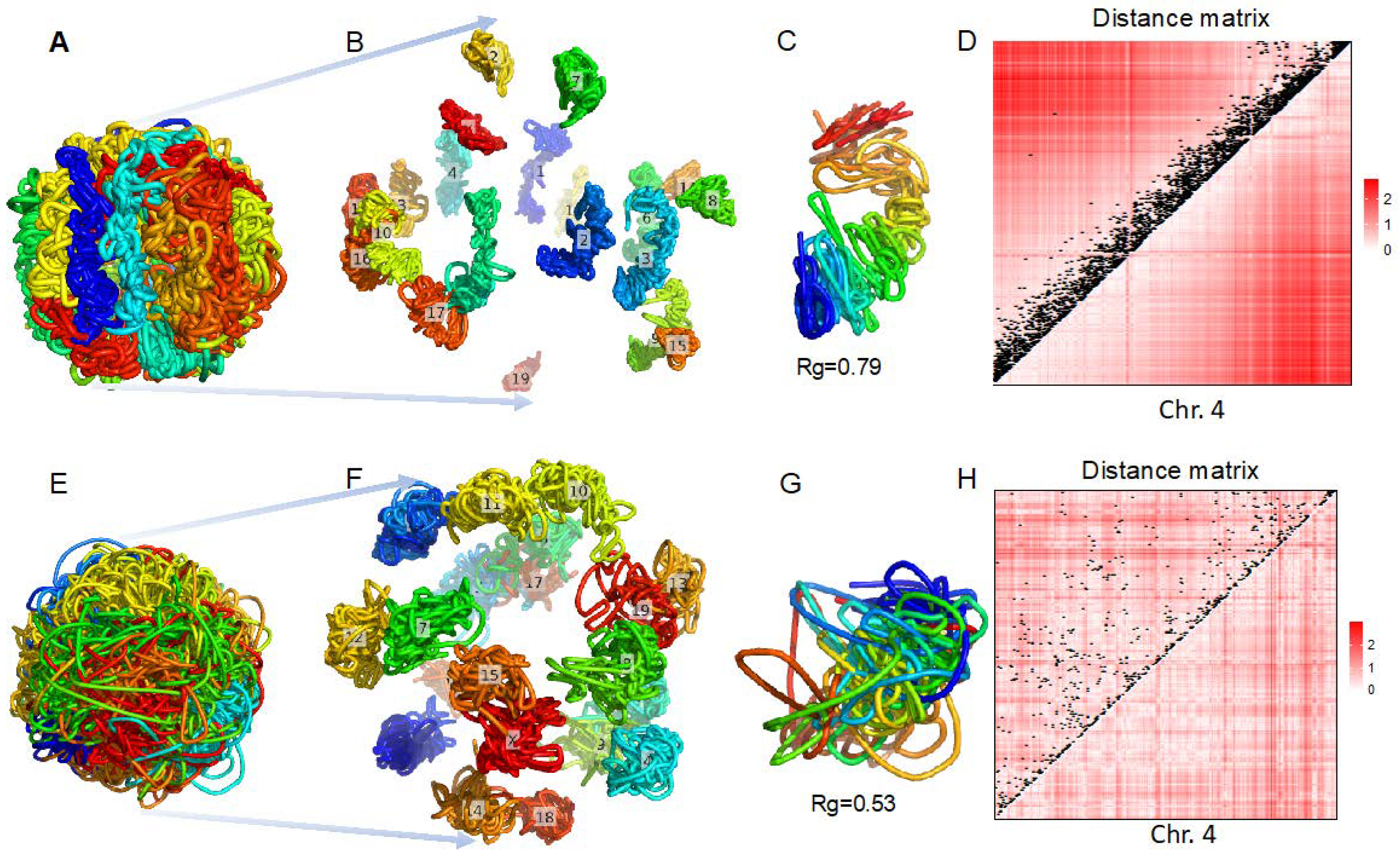
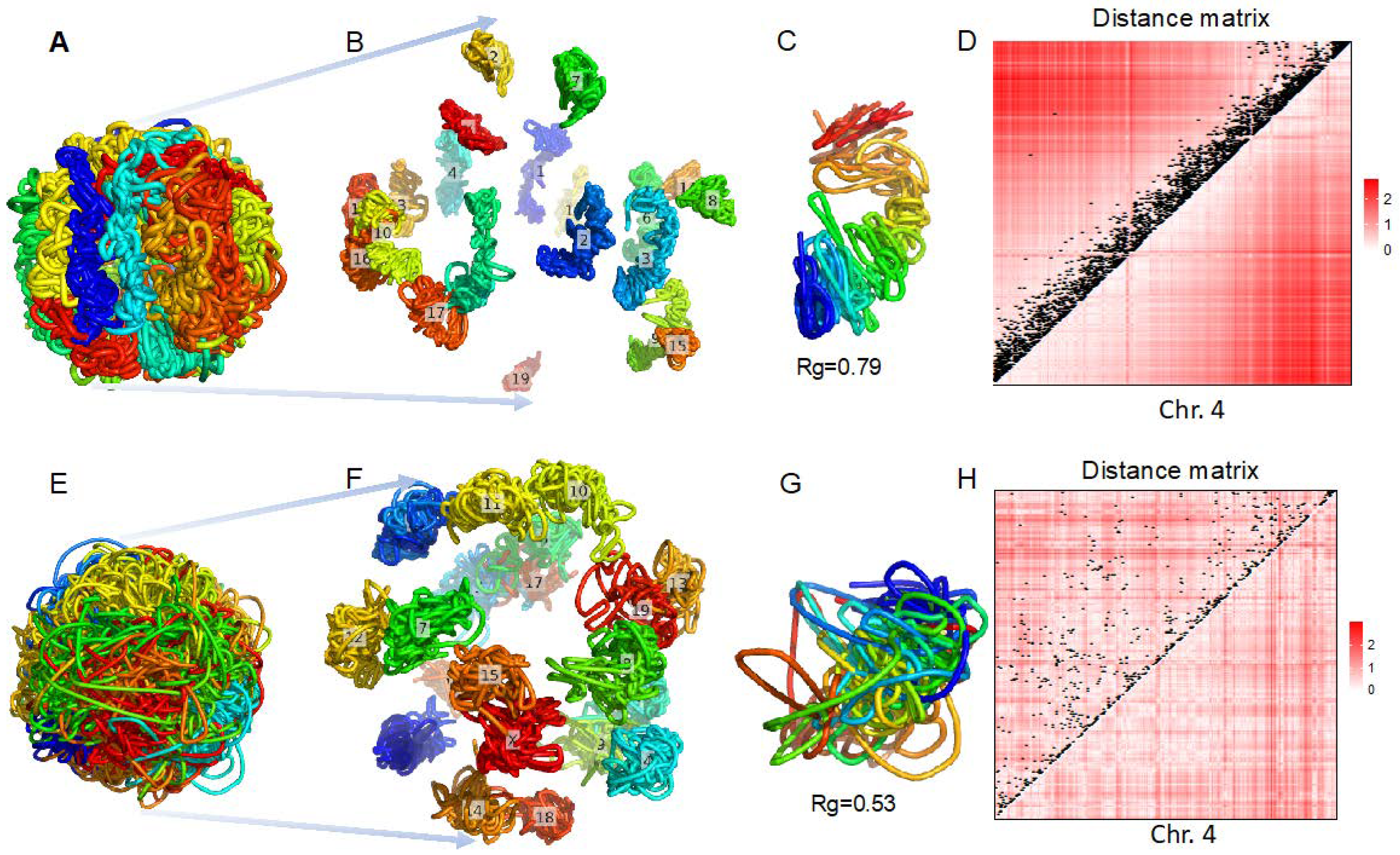
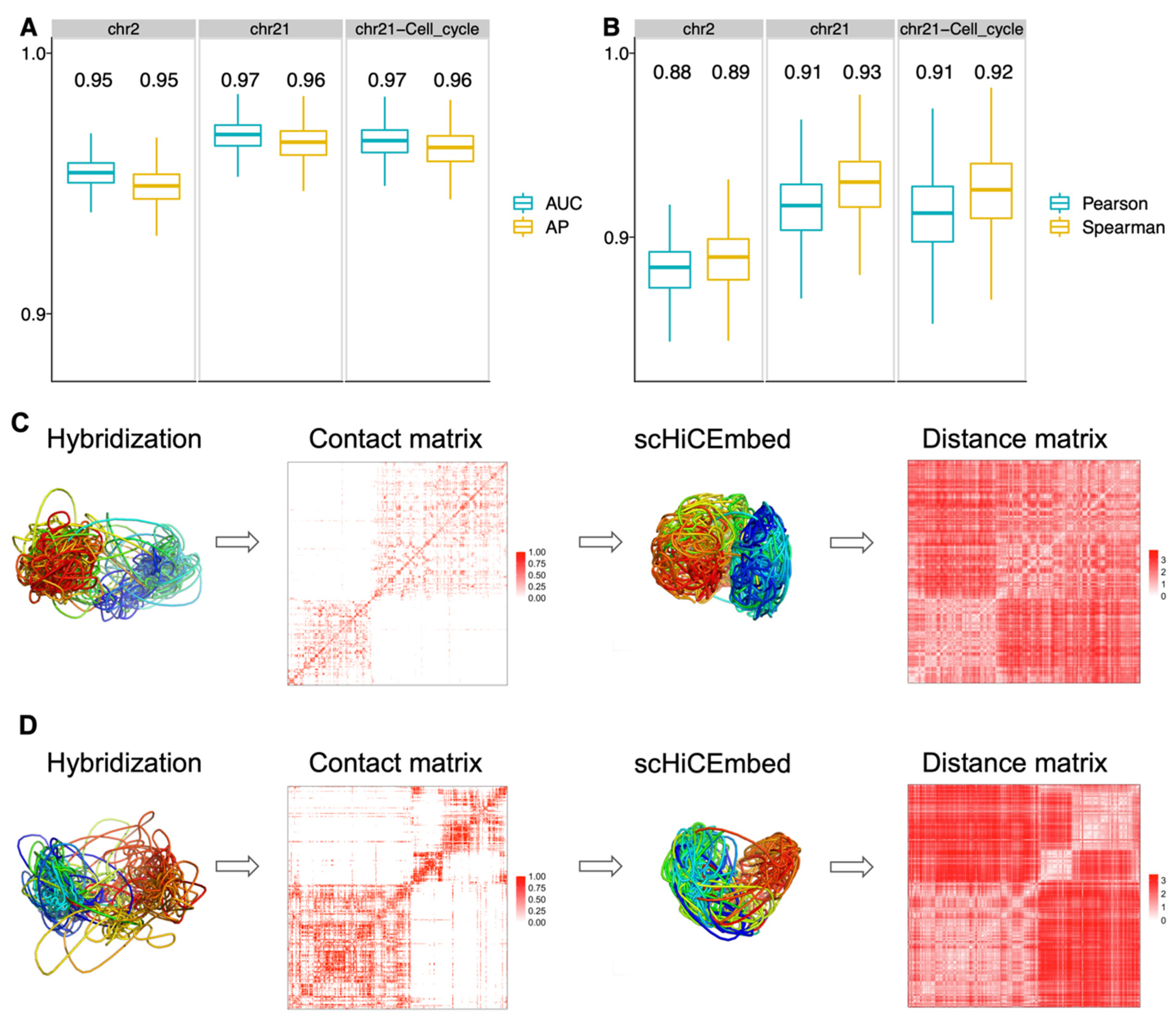
3.2. 3D Genome Structure Reconstruction
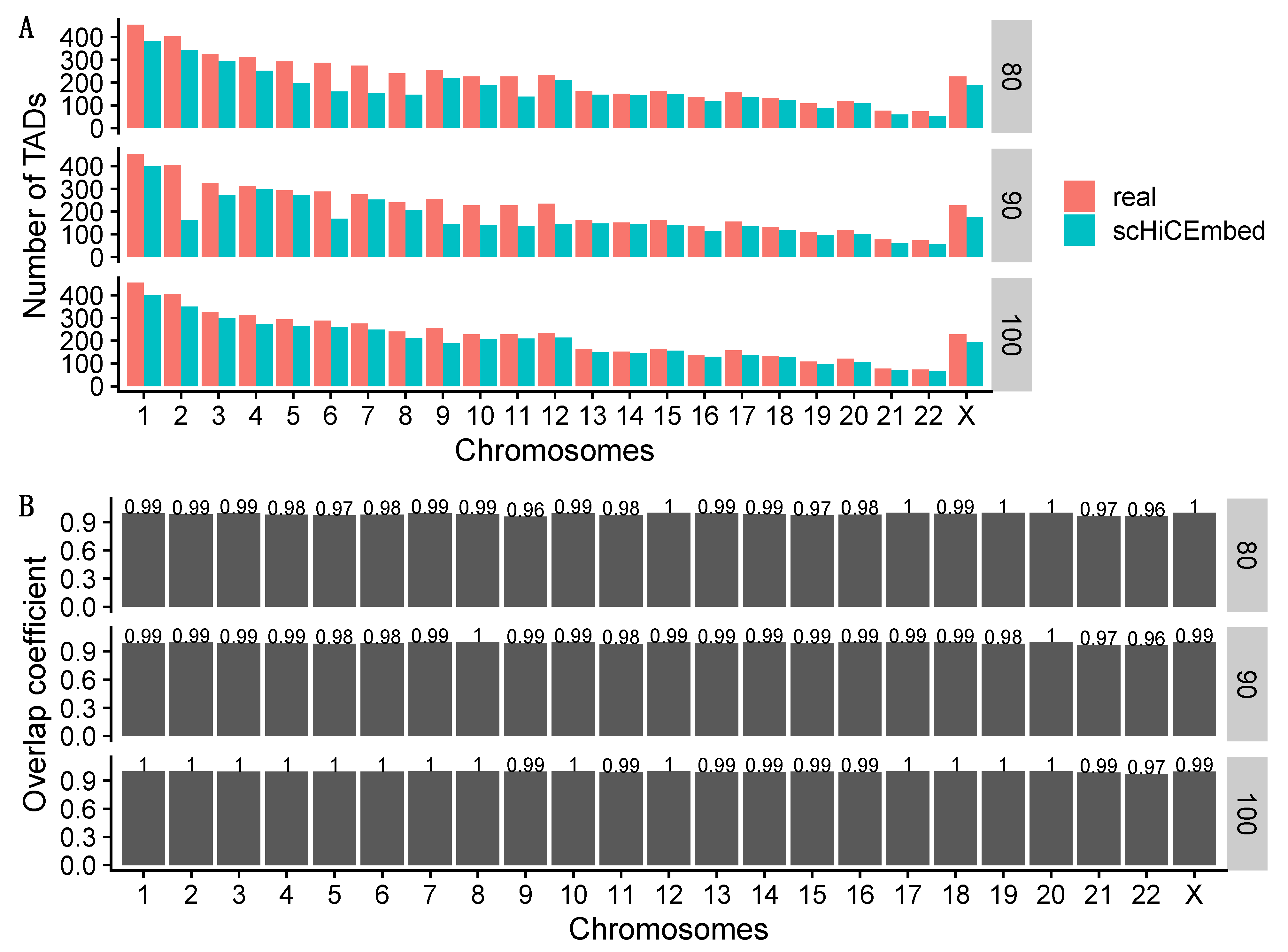
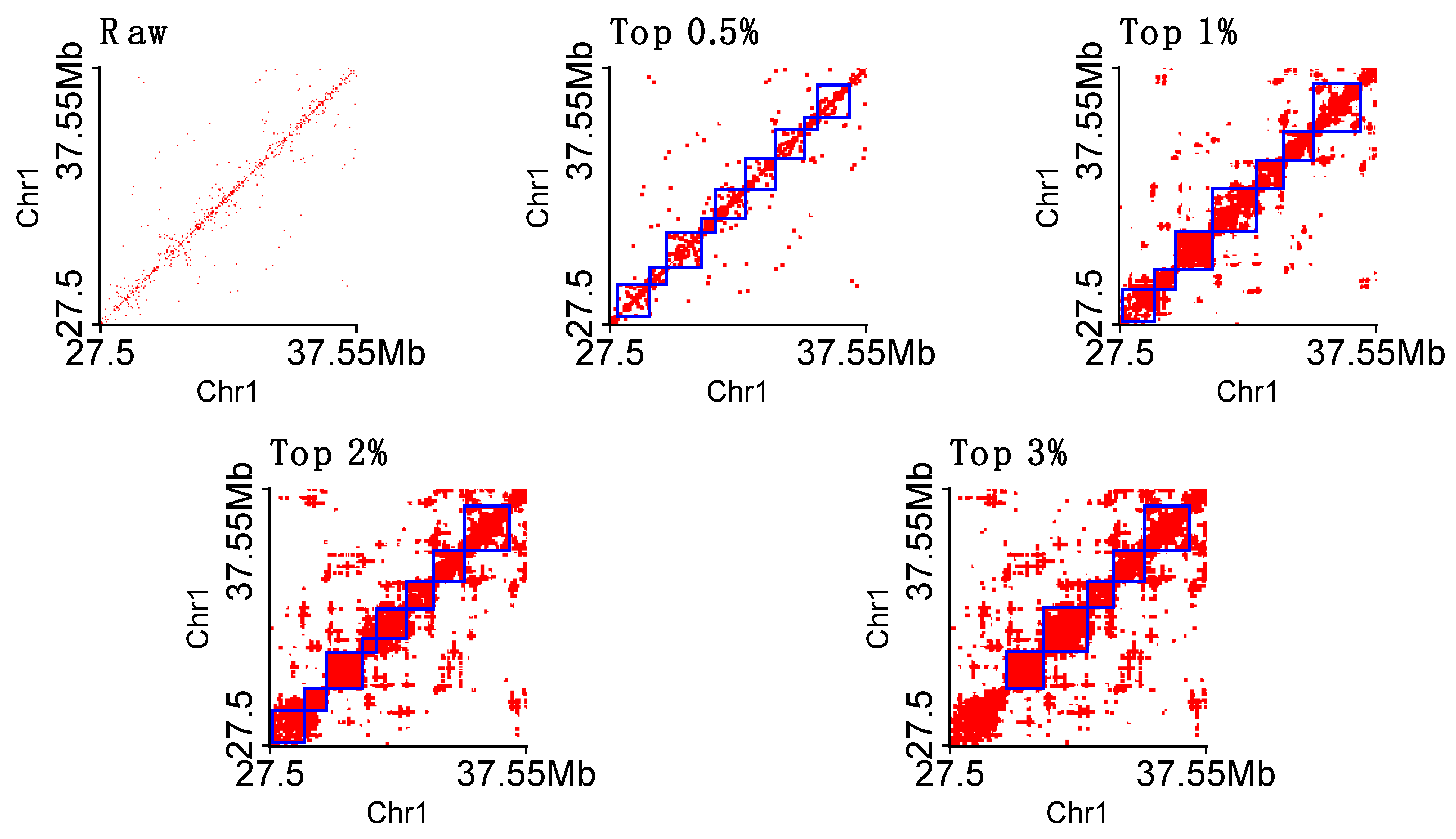
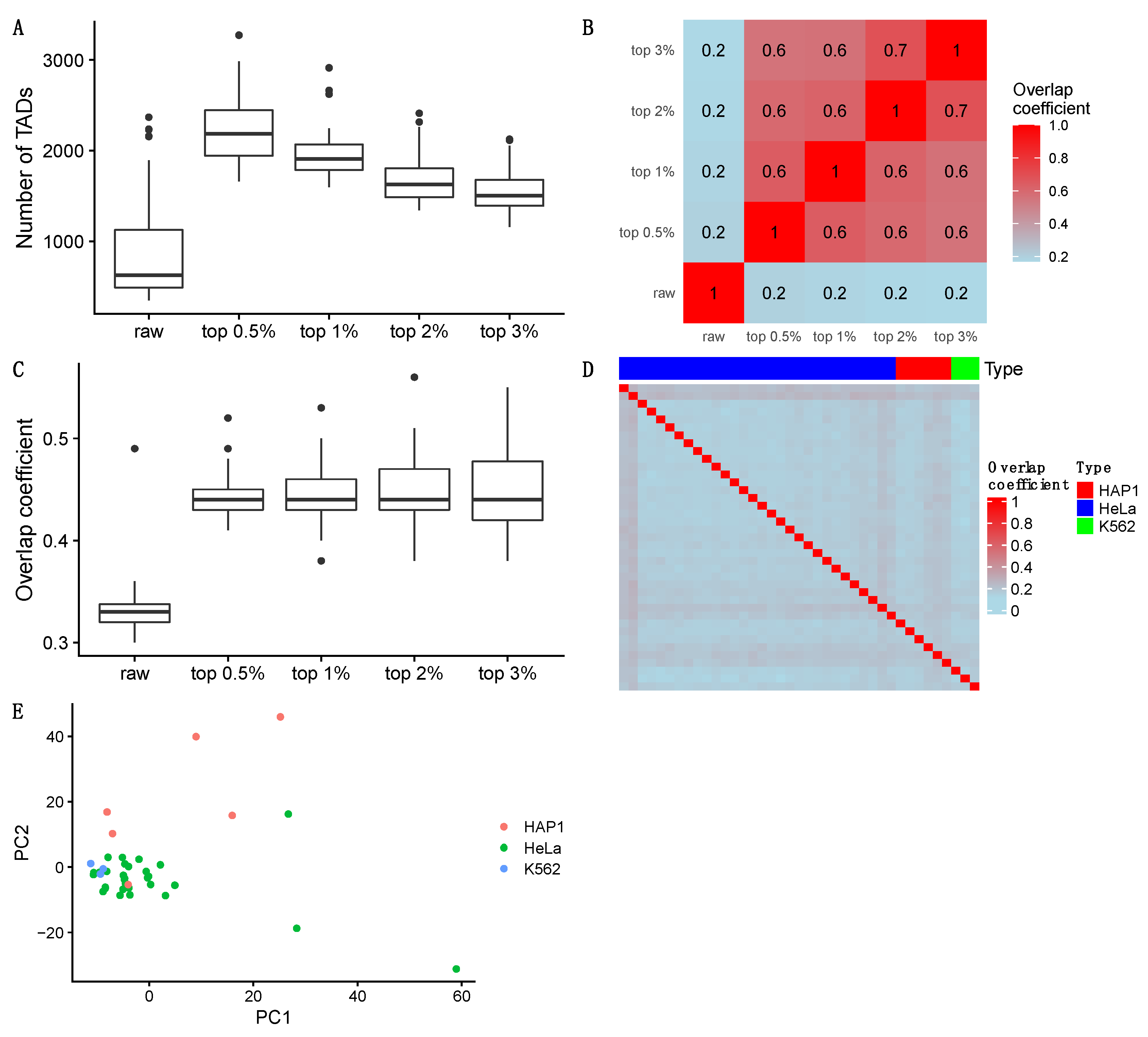
3.3. TAD Detection
3.3.1. Detecting Naïve TADs
3.3.2. Detecting TADs Based on Raw and Imputed Single-Cell Hi-C Data
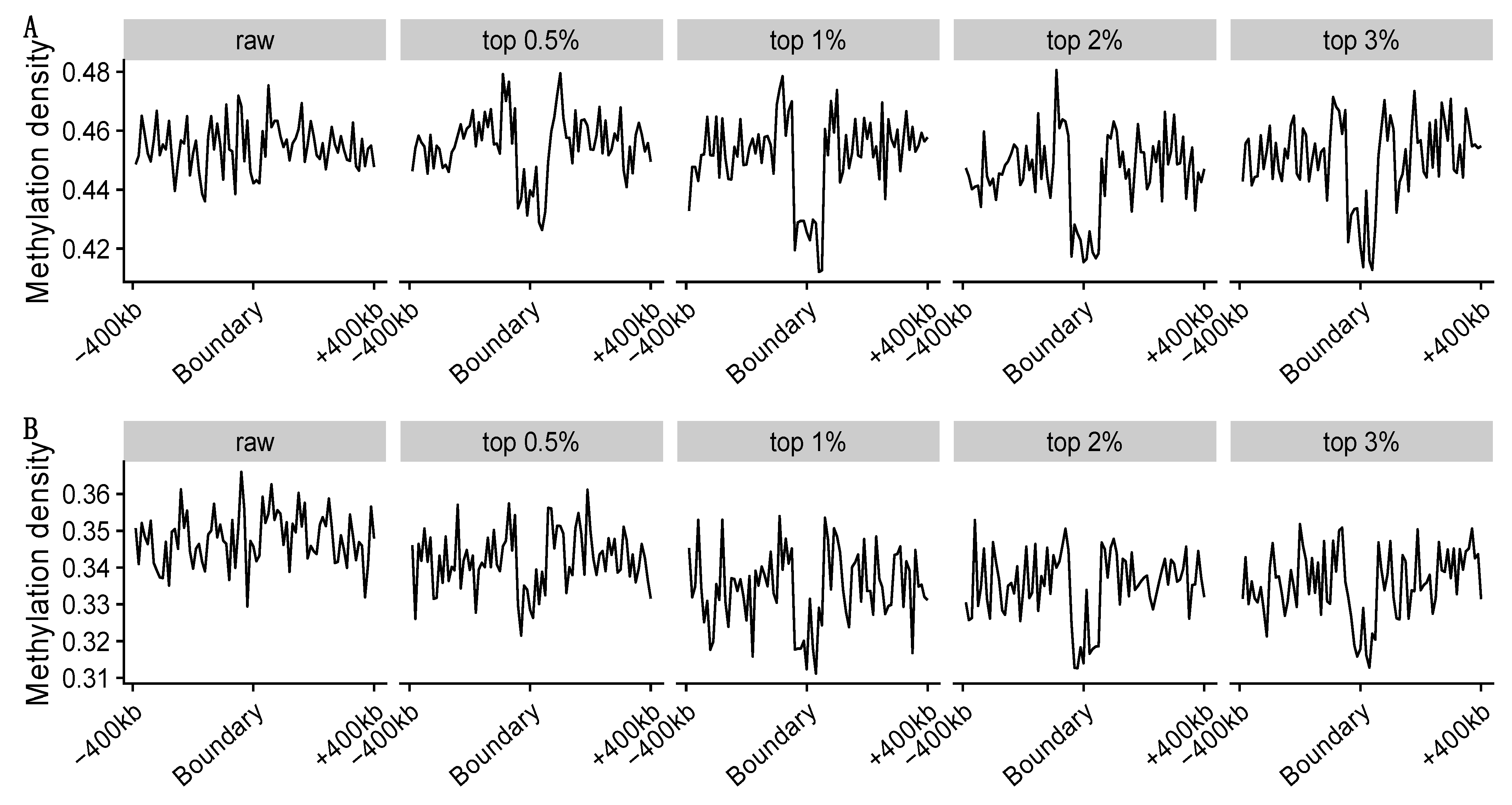
3.3.3. Methylation Loss around the TAD Boundaries
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Ay, F.; Lee, C.; Gulsoy, G.; Deng, X.; Cook, S.; Hesson, J.; Cavanaugh, C.; Ware, C.B.; Krumm, A.; et al. Fine-scale chromatin interaction maps reveal the cis-regulatory landscape of human lincRNA genes. Nat. Methods 2015, 12, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.-H.S.; Weiner, A.; Lajoie, B.; Dekker, J.; Friedman, N.; Rando, O.J. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell 2015, 162, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Mumbach, M.R.; Rubin, A.J.; Flynn, R.A.; Dai, C.; Khavari, P.A.; Greenleaf, W.J.; Chang, H.Y. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 2016, 13, 919–922. [Google Scholar] [CrossRef]
- Dryden, N.H.; Broome, L.R.; Dudbridge, F.; Johnson, N.; Orr, N.; Schoenfelder, S.; Nagano, T.; Andrews, S.; Wingett, S.; Kozarewa, I.; et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res. 2014, 24, 1854–1868. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Hu, M.; Deng, K.; Qin, Z.; Dixon, J.; Selvaraj, S.; Fang, J.; Ren, B.; Liu, J.S. Bayesian inference of spatial organizations of chromosomes. PLoS Comput. Biol. 2013, 9, e1002893. [Google Scholar] [CrossRef]
- Varoquaux, N.; Ay, F.; Noble, W.S.; Vert, J.-P. A statistical approach for inferring the 3D structure of the genome. Bioinformatics 2014, 30, i26–i33. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Z. Measuring the three-dimensional structural properties of topologically associating domains. In Proceedings of the 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Madrid, Spain, 3–6 December 2018; pp. 21–28. [Google Scholar]
- Wang, Y.H.; Liu, T.; Xu, D.; Shi, H.D.; Zhang, C.Y.; Mo, Y.Y.; Wang, Z. Predicting DNA Methylation State of CpG Dinucleotide Using Genome Topological Features and Deep Networks. Sci. Rep. 2016, 6, 19598. [Google Scholar] [CrossRef]
- Bonev, B.; Cohen, N.M.; Szabo, Q.; Fritsch, L.; Papadopoulos, G.L.; Lubling, Y.; Xu, X.; Lv, X.; Hugnot, J.-P.; Tanay, A.; et al. Multiscale 3D genome rewiring during mouse neural development. Cell 2017, 171, 557–572.e24. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Xu, J.; Dileep, V.; Zhan, Y.; Song, F.; Le, V.T.; Yardımcı, G.G.; Chakraborty, A.; Bann, D.V.; Wang, Y.; et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 2018, 50, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Lubling, Y.; Várnai, C.; Dudley, C.; Leung, W.; Baran, Y.; Cohen, N.M.; Wingett, S.; Fraser, P.; Tanay, A. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 2017, 547, 61–67. [Google Scholar] [CrossRef]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 2017, 544, 59. [Google Scholar] [CrossRef]
- Ramani, V.; Deng, X.; Qiu, R.; Gunderson, K.L.; Steemers, F.J.; Disteche, C.M.; Noble, W.S.; Duan, Z.; Shendure, J. Massively multiplex single-cell Hi-C. Nat. Methods 2017, 14, 263–266. [Google Scholar] [CrossRef]
- Flyamer, I.M.; Gassler, J.; Imakaev, M.; Brandão, H.B.; Ulianov, S.V.; Abdennur, N.; Razin, S.V.; Mirny, L.A.; Tachibana-Konwalski, K. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 2017, 544, 110–114. [Google Scholar] [CrossRef]
- Li, G.; Liu, Y.; Zhang, Y.; Kubo, N.; Yu, M.; Fang, R.; Kellis, M.; Ren, B. Joint profiling of DNA methylation and chromatin architecture in single cells. Nat. Methods 2019, 16, 991–993. [Google Scholar] [CrossRef]
- Tan, L.; Xing, D.; Chang, C.-H.; Li, H.; Xie, X.S. Three-dimensional genome structures of single diploid human cells. Science 2018, 361, 924–928. [Google Scholar] [CrossRef]
- Lee, D.S.; Luo, C.; Zhou, J.; Chandran, S.; Rivkin, A.; Bartlett, A.; Nery, J.R.; Fitzpatrick, C.; O’Connor, C.; Dixon, J.R.; et al. Simultaneous profiling of 3D genome structure and DNA methylation in single human cells. Nat. Methods 2019, 16, 999–1006. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, Z. SCL: A lattice-based approach to infer 3D chromosome structures from single-cell Hi-C data. Bioinformatics 2019, 35, 3981–3988. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.E.; Corces, V.G. CTCF: Master weaver of the genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; Xuan, Z.; Chen, M.; Zhang, M.Q. De novo deciphering three-dimensional chromatin interaction and topological domains by wavelet transformation of epigenetic profiles. Nucleic Acids Res. 2016, 44, e106. [Google Scholar] [CrossRef] [PubMed]
- Filippova, D.; Patro, R.; Duggal, G.; Kingsford, C. Identification of alternative topological domains in chromatin. Algorithms Mol. Biol. 2014, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Lévy-Leduc, C.; Delattre, M.; Mary-Huard, T.; Robin, S. Two-dimensional segmentation for analyzing Hi-C data. Bioinformatics 2014, 30, i386–i392. [Google Scholar] [CrossRef] [PubMed]
- Libbrecht, M.W.; Ay, F.; Hoffman, M.M.; Gilbert, D.M.; Bilmes, J.A.; Noble, W.S. Joint annotation of chromatin state and chromatin conformation reveals relationships among domain types and identifies domains of cell-type-specific expression. Genome Res. 2015, 25, 544–557. [Google Scholar] [CrossRef]
- Shin, H.; Shi, Y.; Dai, C.; Tjong, H.; Gong, K.; Alber, F.; Zhou, X.J. TopDom: An efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 2016, 44, e70. [Google Scholar] [CrossRef]
- Weinreb, C.; Raphael, B.J. Identification of hierarchical chromatin domains. Bioinformatics 2016, 32, 1601–1609. [Google Scholar] [CrossRef]
- Crane, E.; Bian, Q.; McCord, R.P.; Lajoie, B.R.; Wheeler, B.S.; Ralston, E.J.; Uzawa, S.; Dekker, J.; Meyer, B.J. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 2015, 523, 240. [Google Scholar] [CrossRef]
- Zhou, J.; Ma, J.; Chen, Y.; Cheng, C.; Bao, B.; Peng, J.; Sejnowski, T.J.; Dixon, J.R.; Ecker, J.R. Robust single-cell Hi-C clustering by convolution- and random-walk–based imputation. Proc. Natl. Acad. Sci. USA 2019, 116, 14011–14018. [Google Scholar] [CrossRef] [PubMed]
- Kipf, T.N.; Welling, M. Variational graph auto-encoders. arXiv 2016, arXiv:1611.07308. [Google Scholar]
- Yang, T.; Zhang, F.; Yardımcı, G.G.; Song, F.; Hardison, R.C.; Noble, W.S.; Yue, F.; Li, Q. HiCRep: Assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res. 2017, 27, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Bousquet, O.; Lal, T.; Weston, J.; Schölkopf, B. Learning with local and global consistency. Adv. Neural Inf. Process. Syst. 2003, 16, 321–328. [Google Scholar]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Nair, V.; Hinton, G.E. Rectified linear units improve restricted boltzmann machines. In Proceedings of the 27th International Conference on Machine Learning (ICML-10), Haifa, Isreal, 21–24 June 2010; pp. 807–814. [Google Scholar]
- Salha, G.; Hennequin, R.; Vazirgiannis, M. Simple and effective graph autoencoders with one-hop linear models. arXiv 2020, arXiv:2001.07614. [Google Scholar]
- Hadsell, R.; Chopra, S.; LeCun, Y. Dimensionality reduction by learning an invariant mapping. In Proceedings of the 2006 IEEE Computer Society Conference on Computer Vision and Pattern Recognition (CVPR’06), New York, NY, USA, 17–22 June 2006; pp. 1735–1742. [Google Scholar]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L. Pytorch: An imperative style, high-performance deep learning library. In Proceedings of the Advances in Neural Information Processing Systems, Vancouver, BC, Canada, 8–14 December 2019; pp. 8026–8037. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Juggins, S. rioja: Analysis of Quaternary Science Data. 2015. Available online: https://cran.r-project.org/web/packages/rioja/index.html (accessed on 28 October 2020).
- Soler-Vila, P.; Cuscó, P.; Farabella, I.; Di Stefano, M.; Marti-Renom, M.A. Hierarchical chromatin organization detected by TADpole. Nucleic Acids Res. 2020, 48, e39. [Google Scholar] [CrossRef]
- Halkidi, M.; Vazirgiannis, M. Clustering validity assessment: Finding the optimal partitioning of a data set. In Proceedings of the 2001 IEEE International Conference on Data Mining, San Jose, CA, USA, 29 November–2 December 2001; pp. 187–194. [Google Scholar]
- Liu, Y.; Li, Z.; Xiong, H.; Gao, X.; Wu, J. Understanding of internal clustering validation measures. In Proceedings of the 2010 IEEE International Conference on Data Mining, Sydney, Australia, 13 December 2010; pp. 911–916. [Google Scholar]
- Su, J.-H.; Zheng, P.; Kinrot, S.S.; Bintu, B.; Zhuang, X. Genome-scale imaging of the 3D organization and transcriptional activity of chromatin. Cell 2020, 182, 1641–1659.e26. [Google Scholar] [CrossRef]
- Bintu, B.; Mateo, L.J.; Su, J.-H.; Sinnott-Armstrong, N.A.; Parker, M.; Kinrot, S.; Yamaya, K.; Boettiger, A.N.; Zhuang, X. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 2018, 362, eaau1783. [Google Scholar] [CrossRef]
- Xie, W.J.; Qi, Y.; Zhang, B. Characterizing chromatin folding coordinate and landscape with deep learning. PLoS Comput. Biol. 2020, 16, e1008262. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405, 482–485. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index | References | Cell Clustering | TAD Detection | 3D Reconstruction | Resolutions | Cell Types |
|---|---|---|---|---|---|---|
| 1 | [18] | ✓ | 1 Mb | Human Oocyte, Zygote-P, and Zygote-M | ||
| 2 | [17] | ✓ | ✓ | 1 Mb and 50 kb | Human HeLa, HAP1, GM12878, and K562 | |
| 3 | [19] | ✓ | 50 kb | Mouse Embryonic stem | ||
| 4 | [15] | ✓ | 1 Mb and 500 kb | Mouse Embryonic stem |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Wang, Z. scHiCEmbed: Bin-Specific Embeddings of Single-Cell Hi-C Data Using Graph Auto-Encoders. Genes 2022, 13, 1048. https://doi.org/10.3390/genes13061048
Liu T, Wang Z. scHiCEmbed: Bin-Specific Embeddings of Single-Cell Hi-C Data Using Graph Auto-Encoders. Genes. 2022; 13(6):1048. https://doi.org/10.3390/genes13061048
Chicago/Turabian StyleLiu, Tong, and Zheng Wang. 2022. "scHiCEmbed: Bin-Specific Embeddings of Single-Cell Hi-C Data Using Graph Auto-Encoders" Genes 13, no. 6: 1048. https://doi.org/10.3390/genes13061048
APA StyleLiu, T., & Wang, Z. (2022). scHiCEmbed: Bin-Specific Embeddings of Single-Cell Hi-C Data Using Graph Auto-Encoders. Genes, 13(6), 1048. https://doi.org/10.3390/genes13061048