
Genome-Wide Identification of m6A Writers, Erasers and Readers in Poplar 84K




Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Identification of m6A Pathway Genes in Poplar 84K
2.3. Phylogenetic Tree, Gene Structure, Conserved Motifs, Promoter Prediction and Protein Interaction Analysis
2.4. Quantitative Real-Time Reverse Transcription-PCR (qRT-PCR)
2.5. Homology Modeling of 3D Structures, Molecular Docking and Protein Docking
2.6. Statistical Methods
3. Results
3.1. Genome-Wide Identification of m6A Pathway Genes in Poplar 84K
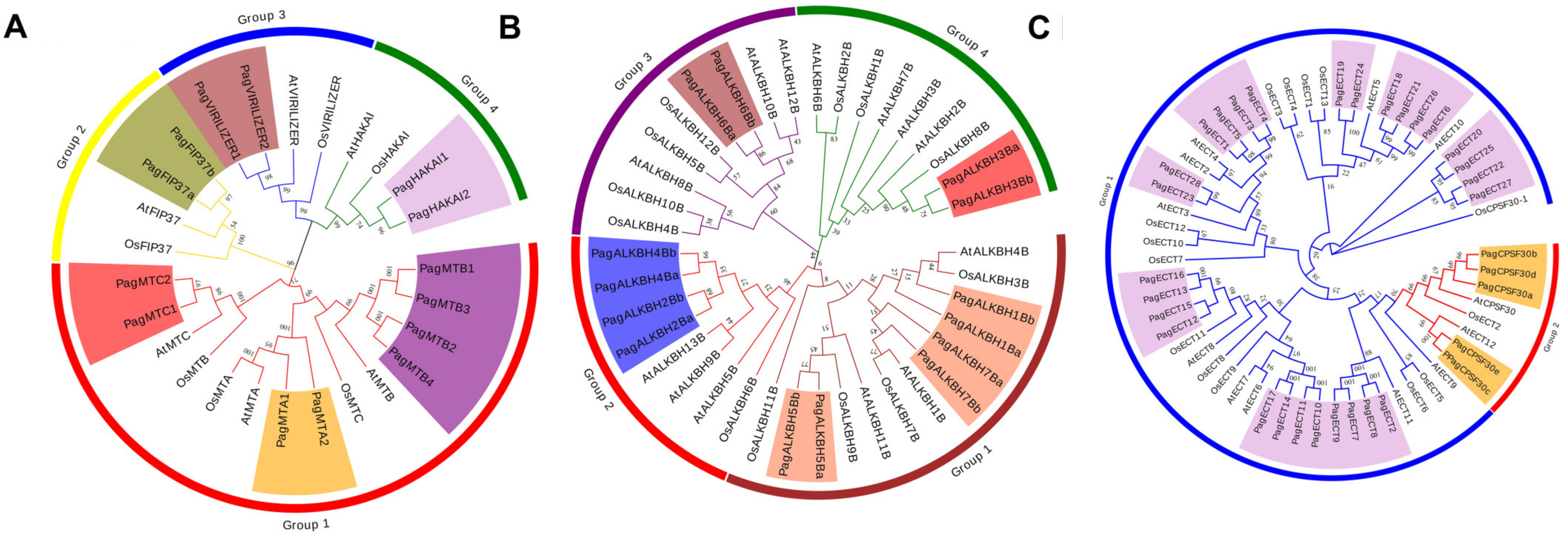
3.2. Phylogenetic Analysis of m6A Pathway Genes in Poplar 84K
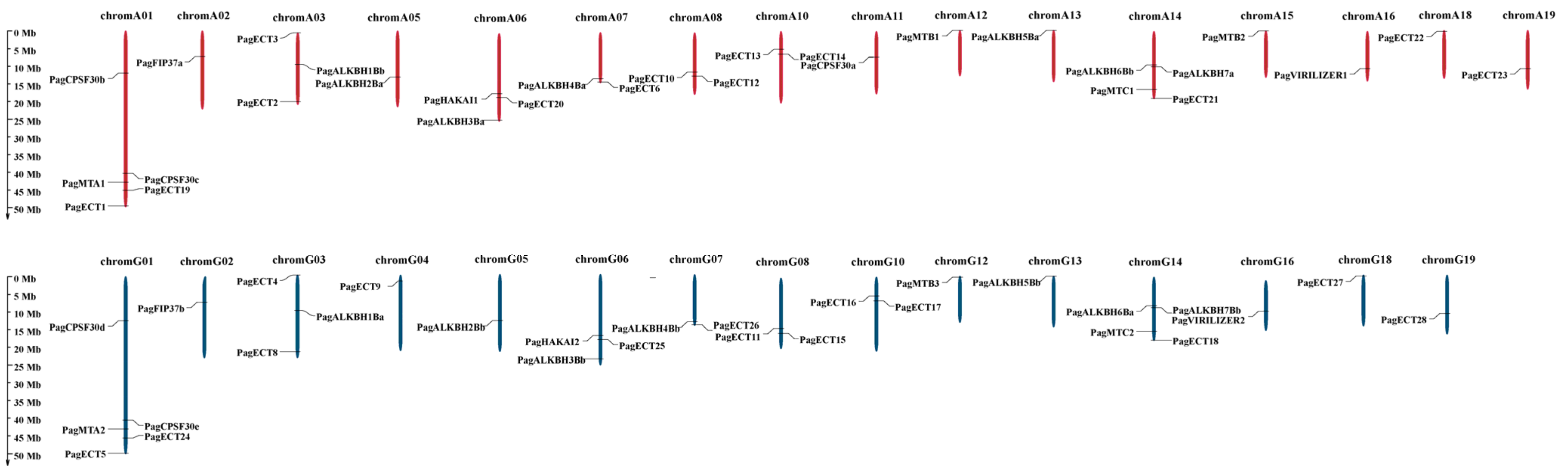
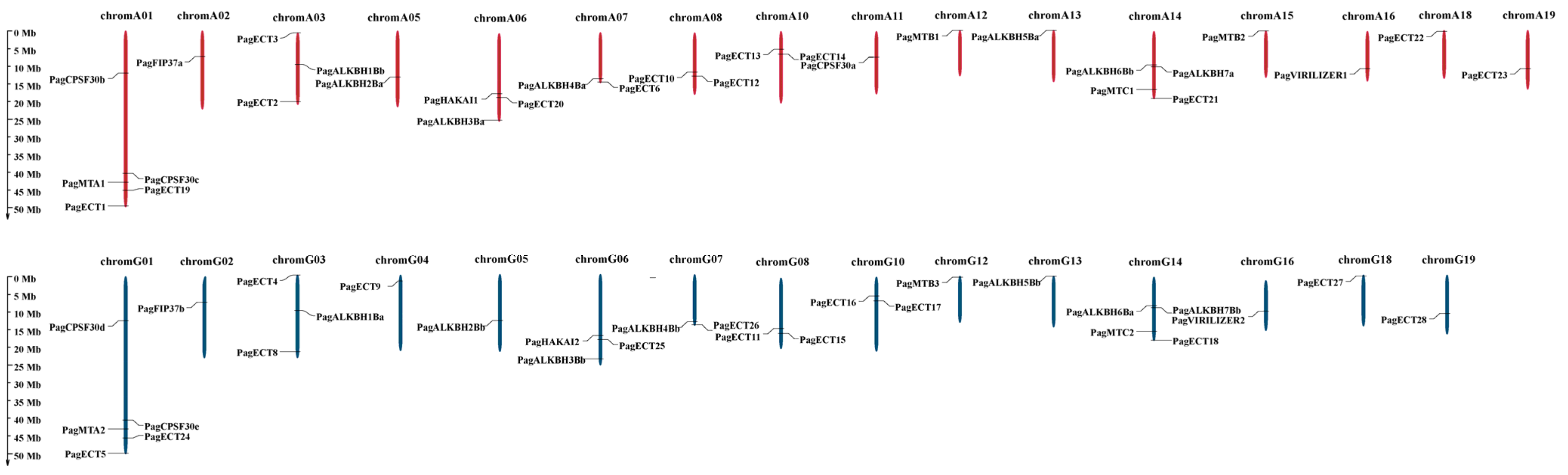
3.3. Analysis of Chromosomal Location
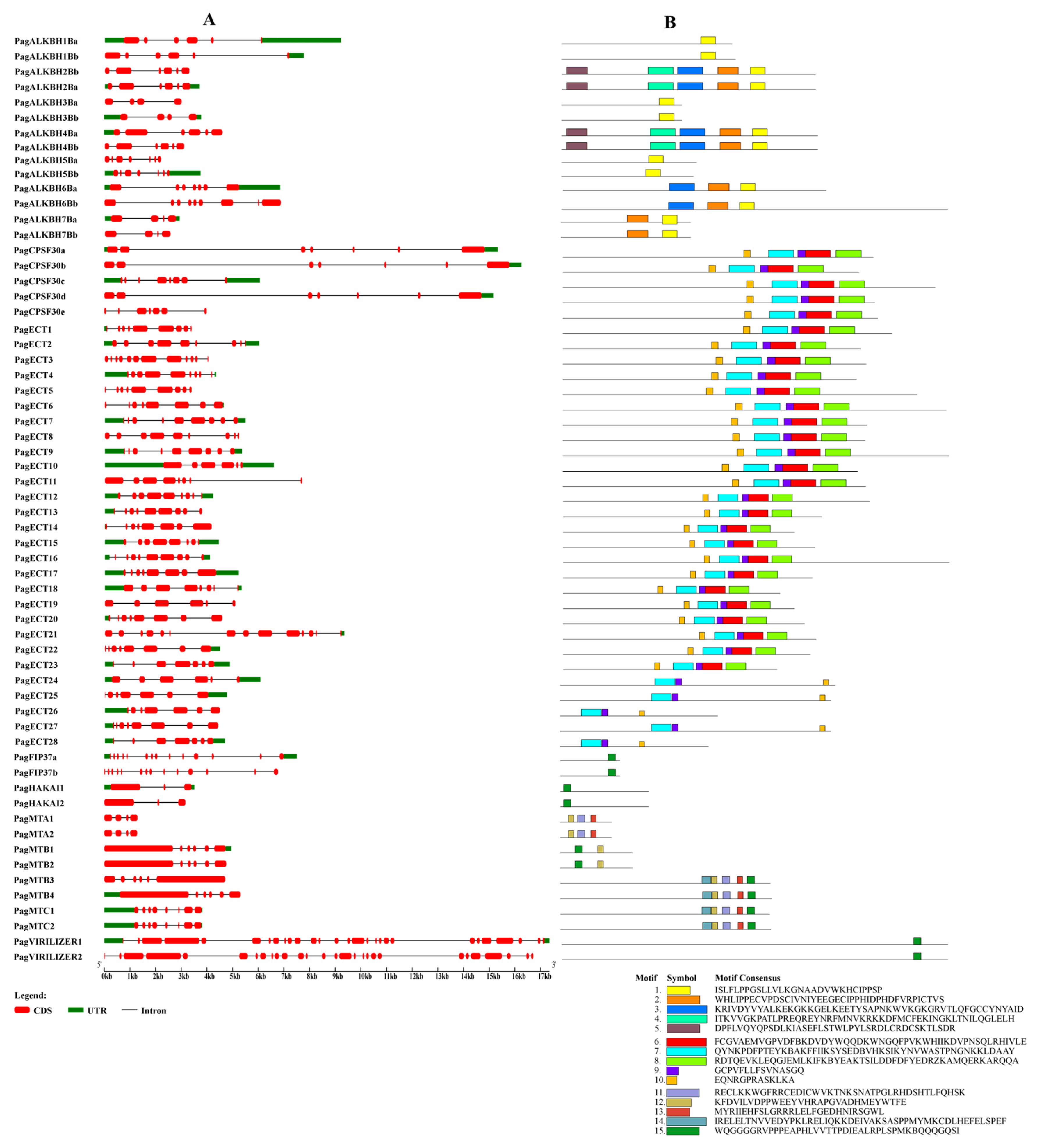
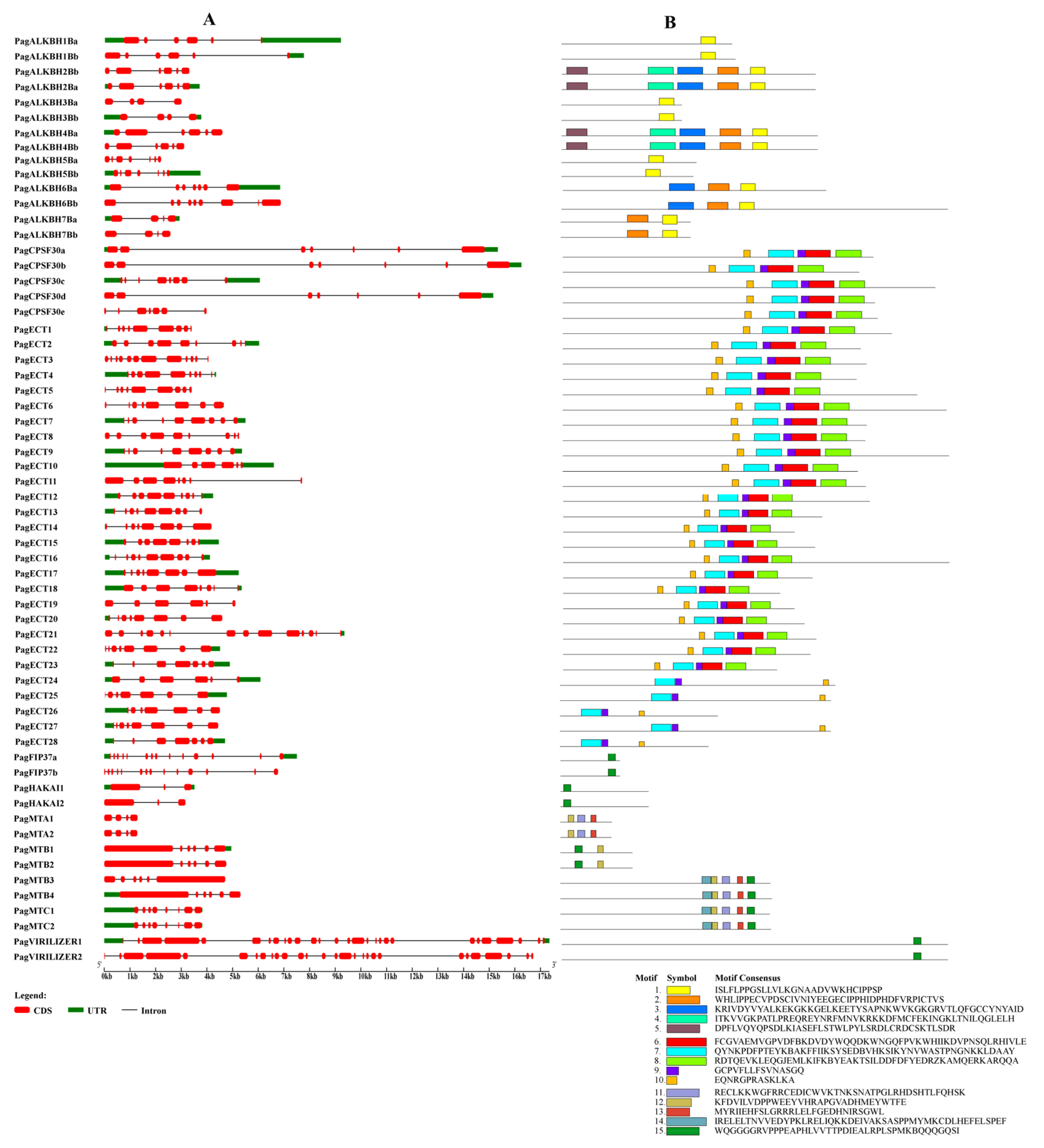
3.4. Gene Structure and Conserved Motif Analysis
3.5. Promoter Analysis of m6A Pathway Genes
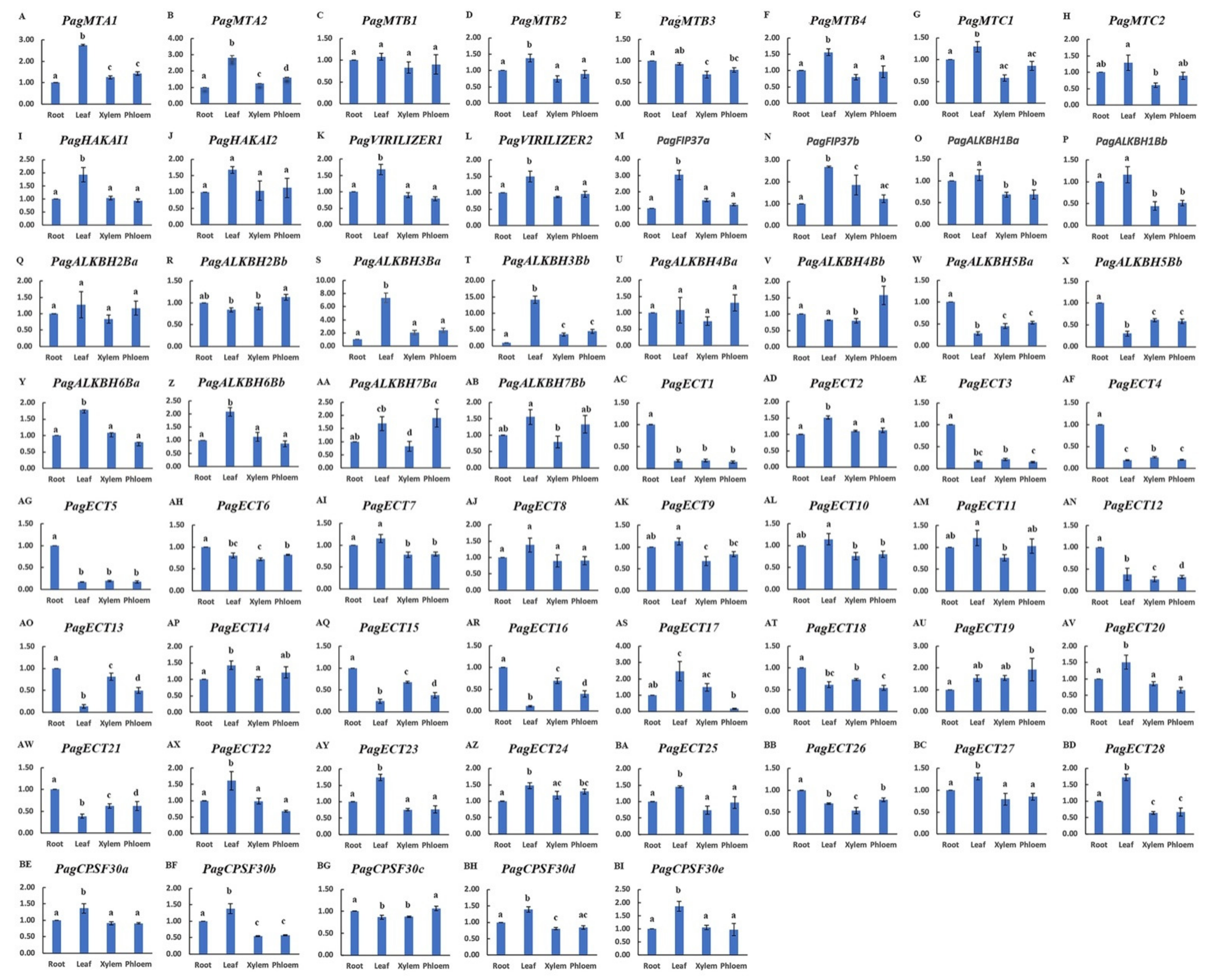
3.6. Expression Profiles of m6A Pathway Genes
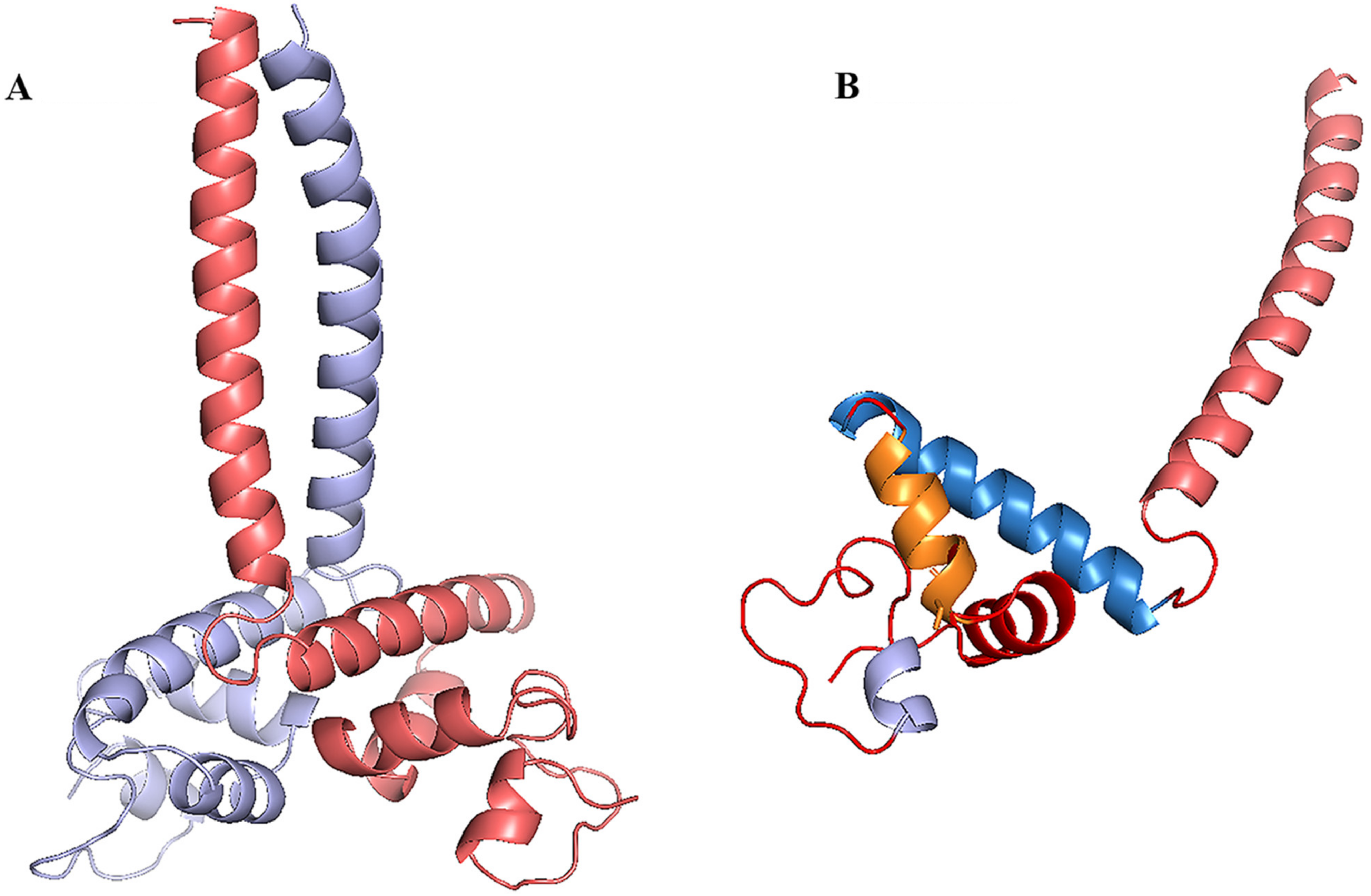
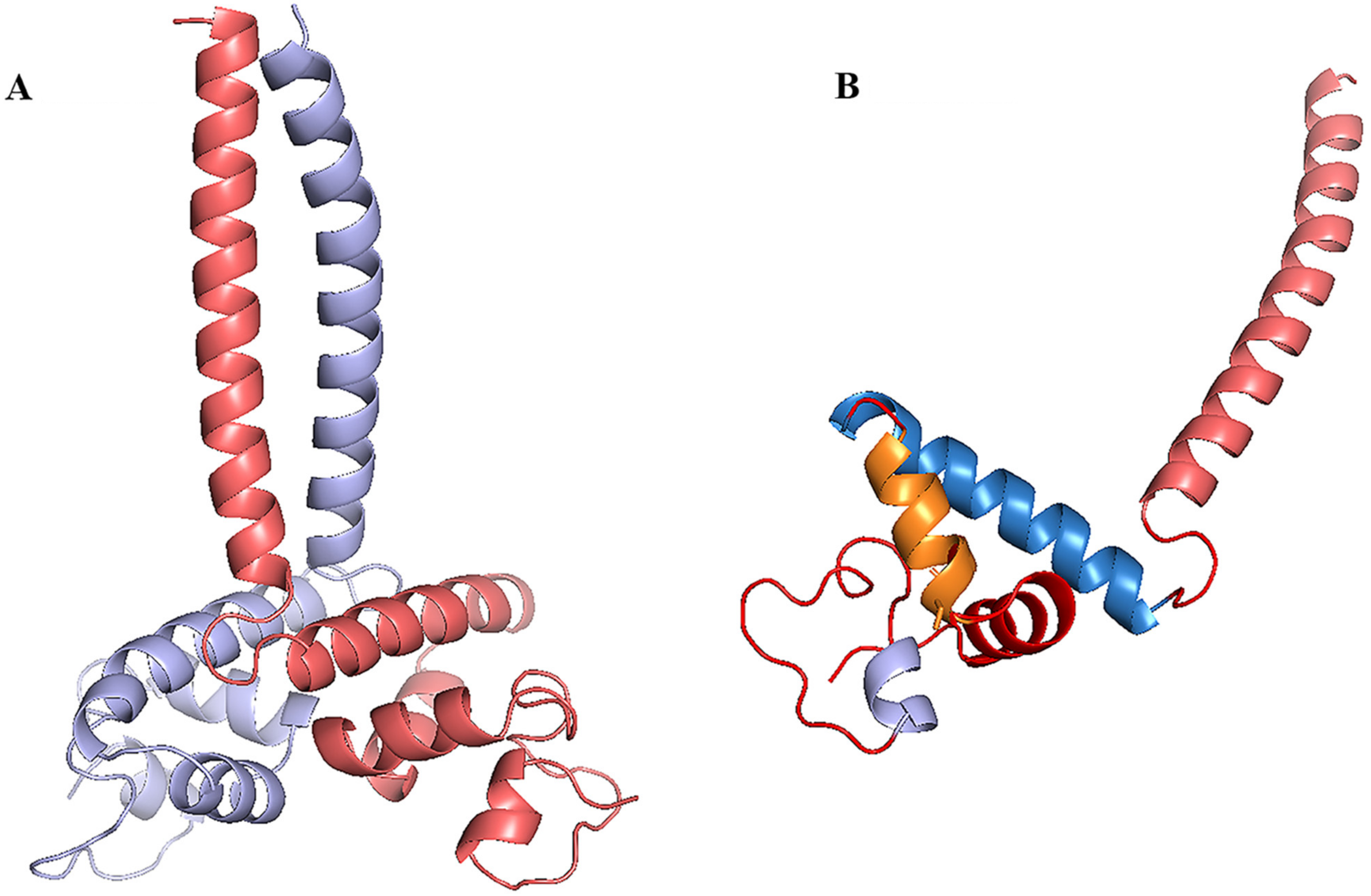
3.7. Homology Modeling, Molecular Docking and Protein Docking
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meyer, K.D.; Jaffrey, S.R. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 2014, 15, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Czerwoniec, A.; Dunin-Horkawicz, S.; Purta, E.; Kaminska, K.H.; Kasprzak, J.M.; Bujnicki, J.M.; Grosjean, H.; Rother, K. MO-DOMICS: A database of RNA modification pathways. 2008 update. Nucleic Acids Res. 2009, 37, 118–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kierzek, E.; Kierzek, R. The thermodynamic stability of RNA duplexes and hairpins containing N6-alkyladenosines and 2-methylthio-N6-alkyladenosines. Nucleic Acids Res. 2003, 31, 4472–4480. [Google Scholar] [CrossRef]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef]
- Arribas-Hernández, L.; Bressendorff, S.; Hansen, M.H.; Poulsen, C.; Erdmann, S.; Brodersen, P. An m6A-YTH Module Controls Developmental Timing and Morphogenesis in Arabidopsis. Plant Cell 2018, 30, 952–967. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.D. m6A-mediated translation regulation. Bbagrm 2018, 1862, 301–309. [Google Scholar] [CrossRef]
- Liu, N.; Pan, T. N6-methyladenosine–encoded epitranscriptomics. Nat. Struct. Mol. Biol. 2016, 23, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Pérez, M.; Aparicio, F.; López-Gresa, M.P.; Bellés, J.M.; Sánchez-Navarro, J.A.; Pallás, V. Arabidopsis m6A demethylase activity modulates viral infection of a plant virus and the m6A abundance in its genomic RNAs. Proc. Natl. Acad. Sci. USA 2017, 114, 10755–10760. [Google Scholar] [CrossRef] [Green Version]
- Duan, H.-C.; Wei, L.-H.; Zhang, C.; Wang, Y.; Chen, L.; Lu, Z.; Chen, P.R.; He, C.; Jia, G. ALKBH10B is An RNA N6-Methyladenosine Demethylase Affecting Arabidopsis Floral Transition. Plant Cell 2017, 29, 2995–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, S.; Li, H.; Bodi, Z.; Button, J.; Vespa, L.; Herzog, M. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 2008, 20, 1278–1288. [Google Scholar] [CrossRef] [Green Version]
- Nichols, J.L. ‘Cap’ structures in maize poly(A)-containing RNA. Biochim. Biophys. Acta 1979, 563, 490–495. [Google Scholar] [CrossRef]
- Kennedy, T.D.; Lane, B.G. Wheat embryo ribonucleates. XIII. Methyl-substituted nucleoside constituents and 5ʹ-terminal dinucleotide sequences in bulk poly (AR)-rich RNA from imbibing wheat embryos. Can. J. Biochem. Cell Biol. 1979, 57, 927–931. [Google Scholar]
- Haugland, R.A.; Cline, M.G. Post-transcriptional modifications of oat coleoptile ribonucleic acids: 5ʹ-terminal capping and methylation of internal nucleosides in poly(A)-rich RNA. Eur. J. Biochem. 1980, 104, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Li, C.; Hu, S.; Yu, J.; Song, S. Transcriptome-wide N6-methyladenosine profiling of rice callus and leaf reveals the presence of tissue-specific competitors involved in selective mRNA modification. RNA Biol. 2014, 11, 1180–1188. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Lv, Z.; Diao, S.; Liu, H.; Duan, A.; He, C.; Zhang, J. Unique features of the m6A methylome and its response to drought stress in sea buckthorn (Hippophae rhamnoides Linn.). RNA Biol. 2021, 18, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Hou, N.; Liu, Z.; He, J. Profiling of N6-Methyladenosine (m6A) Modification Landscape in Response to Drought Stress in Apple (Malus prunifolia (Willd.) Borkh). Plants 2021, 11, 103. [Google Scholar] [CrossRef]
- Bodi, Z.; Zhong, S.; Mehra, S.; Song, J.; Graham, N.; Li, H.; May, S.; Fray, R.G. Adenosine methylation in Arabidopsis mRNA is associated with the 3’ end and reduced levels cause developmental defects. Front. Plant Sci. 2012, 3, 48. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Liang, Z.; Gu, X.; Chen, Y.; Teo, Z.W.N.; Hou, X.; Cai, W.M.; Dedon, P.C.; Liu, L.; Yu, H. N6-Methyladenosine RNA Modification Regulates Shoot Stem Cell Fate in Arabidopsis. Dev. Cell 2016, 38, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Tang, K.; Zhang, D.; Xie, S.; Zhu, X.; Wang, Z.; Lang, Z. Transcriptome-wide high-through put deep m6A-seq reveals unique differential m6A methylation patterns between three organs in Arabidopsis thaliana. Genome Biol. 2015, 16, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Shao, F.J.; Sun, X.C.; Wu, W.L.; Lu, Q.; Wilson, I.W.; Qiu, D.Y. A comparative analysis of differential N6-methyladenosine (m6A) modification between non-transgenic and LBD15 overexpressing Poplar 84K plants. Tree Genet. Genomes 2021, 17, 39. [Google Scholar] [CrossRef]
- Růžička, K.; Zhang, M.; Campilho, A.; Bodi, Z.; Kashif, M.; Saleh, M.; Eeckhout, D.; El-Showk, S.; Li, H.; Zhong, S.; et al. Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 2017, 215, 157–172. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Shao, F.J.; Macmillan, C.; Wilson, I.W.; Merwe, K.; Hussey, S.G.; Myburg, A.A.; Dong, X.; Qiu, D. Genome-wide analysis of the lateral organ boundaries domain gene family in Eucalyptus grandis reveals members that differentially impact secondary growth. Plant Biotechnol. J. 2018, 16, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein detabase search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, 265–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Profiti, G.; Casadio, R. BUSCA: An integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018, 46, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Jiangtao, C.; Yingzhen, K.; Qian, W.; Yuhe, S.; Daping, G.; Jing, L.; Guanshan, L. MapGene2Chrom, a tool to draw gene physical map based on Perl and SVG languages. Yi Chuan 2015, 37, 91–97. [Google Scholar]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene’ feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, 202–208. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Li, Y.Y.; Yang, Y.F.; Wang, S.; Liu, H.; Qiu, D.Y. Cloning and Expression of Lateral Organ Boundaries Domain Genes (TcLBDs) in Taxus Chinensis. Sci. Silvae Sin. 2015, 51, 126–133. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, 296–303. [Google Scholar] [CrossRef] [Green Version]
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Lemma, R.B.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Pérez, N.M.; et al. JASPAR 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022, 50, 165–173. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK Server: Interactive Docking Prediction of Protein-Protein Complexes and Symmetric Multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. ‘Protein interfaces, surfaces and assemblies’ service PISA at the European Bioinformatics Institute. (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) ‘Inference of macromolecular assemblies from crystalline state’. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Schiffrin, B.; Radford, S.E.; Brockwell, D.J.; Calabrese, A.N. PyXlinkViewer: A flexible tool for visualization of protein chemical crosslinking data within the PyMOL molecular graphics system. Protein Sci. 2020, 29, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.S.; Bielewicz, D.; Jarmolowski, A.; Szweykowska-Kulinska, Z. N6-methyladenosine (m6A): Revisiting the Old with Focus on New, an Arabidopsis thaliana Centered Review. Genes 2018, 30, 596. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Length (bp) | Length (aa) | PI | MW (kDa) | Subcellular Location |
|---|---|---|---|---|---|
| PagALKBH1Ba | 6018 | 340 | 6.41 | 37.91 | nucleus |
| PagALKBH1Bb | 7182 | 346 | 6.51 | 38.59 | nucleus |
| PagALKBH2Ba | 3684 | 506 | 5.62 | 56.68 | nucleus |
| PagALKBH2Bb | 3280 | 506 | 5.72 | 56.74 | nucleus |
| PagALKBH3Ba | 2086 | 240 | 9.17 | 27.97 | nucleus |
| PagALKBH3Bb | 2646 | 240 | 9.22 | 28.03 | nucleus |
| PagALKBH4Ba | 3221 | 511 | 6.08 | 57.38 | nucleus |
| PagALKBH4Bb | 3091 | 511 | 5.98 | 57.48 | nucleus |
| PagALKBH5Ba | 2190 | 269 | 5.45 | 30.53 | nucleus |
| PagALKBH5Bb | 3731 | 263 | 5.37 | 29.96 | nucleus |
| PagALKBH6Ba | 6847 | 525 | 5.72 | 56.96 | nucleus |
| PagALKBH6Bb | 6870 | 770 | 6 | 84.95 | nucleus |
| PagALKBH7Ba | 2029 | 259 | 4.62 | 29.28 | nucleus |
| PagALKBH7Bb | 1783 | 259 | 4.7 | 29.33 | nucleus |
| PagFIP37a | 7503 | 336 | 5.22 | 38.04 | nucleus |
| PagFIP37b | 6770 | 336 | 5.22 | 38.08 | nucleus |
| PagHAKAI1 | 3511 | 498 | 6.06 | 53.75 | nucleus |
| PagHAKAI2 | 3162 | 498 | 6.15 | 53.74 | nucleus |
| PagMTA1 | 1294 | 454 | 6.24 | 49.99 | nucleus |
| PagMTA2 | 1285 | 288 | 5.54 | 32.56 | nucleus |
| PagMTB1 | 4948 | 1185 | 7.18 | 131.99 | nucleus |
| PagMTB2 | 4755 | 1193 | 7.4 | 132.93 | nucleus |
| PagMTB3 | 4711 | 1181 | 7.4 | 131.55 | nucleus |
| PagMTB4 | 5307 | 1188 | 7.18 | 132.34 | nucleus |
| PagMTC1 | 3822 | 406 | 6.58 | 46.93 | nucleus |
| PagMTC2 | 3812 | 406 | 6.77 | 46.94 | nucleus |
| PagVIRILIZER1 | 17,347 | 2179 | 5.26 | 238.44 | nucleus |
| PagVIRILIZER2 | 16,715 | 2179 | 5.28 | 23.83 | nucleus |
| PagECT1 | 3401 | 616 | 6.9 | 68.49 | nucleus |
| PagECT2 | 6030 | 588 | 6.25 | 64.73 | nucleus |
| PagECT3 | 4030 | 739 | 8.67 | 81.95 | nucleus |
| PagECT4 | 4325 | 619 | 7.17 | 68.65 | nucleus |
| PagECT5 | 3379 | 625 | 6.44 | 69.20 | nucleus |
| PagECT6 | 4628 | 653 | 5.48 | 71.62 | nucleus |
| PagECT7 | 5476 | 591 | 6.48 | 65.24 | nucleus |
| PagECT8 | 5224 | 602 | 5.94 | 66.12 | nucleus |
| PagECT9 | 5342 | 583 | 7.21 | 64.51 | nucleus |
| PagECT10 | 6578 | 703 | 6.34 | 78.35 | nucleus |
| PagECT11 | 7680 | 761 | 6.1 | 84.21 | nucleus |
| PagECT12 | 4211 | 603 | 5.72 | 66.29 | nucleus |
| PagECT13 | 3780 | 600 | 6.16 | 66.44 | nucleus |
| PagECT14 | 4156 | 766 | 5.95 | 84.53 | nucleus |
| PagECT15 | 4431 | 585 | 5.71 | 64.22 | nucleus |
| PagECT16 | 4089 | 601 | 6.22 | 66.33 | nucleus |
| PagECT17 | 5212 | 774 | 5.98 | 85.56 | nucleus |
| PagECT18 | 5324 | 654 | 5.33 | 71.68 | nucleus |
| PagECT19 | 5080 | 584 | 6.6 | 64.66 | nucleus |
| PagECT20 | 4567 | 636 | 5.2 | 69.39 | nucleus |
| PagECT21 | 9328 | 975 | 6.78 | 108.47 | nucleus |
| PagECT22 | 4478 | 629 | 5.54 | 68.75 | nucleus |
| PagECT23 | 4856 | 548 | 6.58 | 60.02 | nucleus |
| PagECT24 | 6050 | 584 | 6.68 | 64.65 | nucleus |
| PagECT25 | 4745 | 609 | 5.34 | 66.39 | cytoplasm |
| PagECT26 | 4476 | 639 | 5.43 | 70.22 | nucleus |
| PagECT27 | 4409 | 624 | 5.38 | 68.26 | nucleus |
| PagECT28 | 4672 | 540 | 6.25 | 59.01 | cytoplasm |
| PagCPSF30a | 15,333 | 695 | 6.08 | 76.07 | nucleus |
| PagCPSF30b | 16,254 | 684 | 6.2 | 75.03 | nucleus |
| PagCPSF30c | 6063 | 398 | 6.17 | 44.82 | nucleus |
| PagCPSF30d | 15,158 | 684 | 6.2 | 74.99 | nucleus |
| PagCPSF30e | 3987 | 375 | 5.82 | 42.11 | nucleus |
| Receptor Protein | Ligand | Predicted Cis-Element Sequence | Affinity (kcal/mol) | Amino Acid Residues in Docking | |
|---|---|---|---|---|---|
| A Chain | B Chain | ||||
| LBD15 | PagFIP37b | CACCCGGAATTT | −5.3 | Thr-44 Cys-56 Asn-103 Tyr-107 Arg-114 | |
| LBD15 | PagALKBH4Ba | AAACCGGAAAAG | −5 | Arg-53 | |
| LBD15 | PagECT2 | AAGCAGGAACTT | −4.8 | Gln-129 | Gln-128 Gln-132 |
| LBD15 | PagHAKAI2 | CAGCAGGAGACA | −4.8 | Arg-55 Ser-84 | |
| LBD15 | PagCPSF30a | CCCCCAGAAAAT | −4.7 | Pro-45 Arg-55 Cys-56 Gln-58 Ser-63 Pro-64 Asp-100 Asn-103 | |
| LBD15 | PagALKBH2Ba | CTGCCTGAGACA | −4.6 | Leu-52 Arg-55 Glu-95 | |
| LBD15 | PagECT23 | TCGCAGGCAATG | −4.5 | Try-107 Asn-110 | Gln-128 |
| LBD15 | PagECT13 | GAGCTGGAAAAT | −4.4 | Tyr-107 | Asn-139 Ser-143 |
| LBD15 | PagECT15 | GTTCCACCACCTG | −4.3 | Glu-70 Asn-103 Aan-110 | Ser-125 |
| LBD15 | PagECT28 | TCGCAGGCAATG | −4.1 | Thr-44 Glu-59 Cys-60 Lys-73 Ser-84 | |
| LBD15 | PagECT9 | TCTCAGGAAACA | −4.1 | Lys-73 Asn-110 Arg-114 | |
| LBD15 | PagECT16 | TCTCCGCCGTCCC | −4.0 | Phe-80 Ser-84 | His-78 Asn-85 |
| LBD15 | PagECT7 | AAACCGGAATAT | −3.7 | His-69 Lys-73 Asn-103 | Gln-128 |
| Structure 1 | Structure 2 | Interface | Inte | Inte | NHB | NSB |
|---|---|---|---|---|---|---|
| Area, Å2 | kcal/M | p-Value | ||||
| PagMTA1 | PagMTC1 | 3151.8 | −18.3 | 0.471 | 34 | 12 |
| PagMTA1 | PagMTC2 | 2655.1 | −9.2 | 0.552 | 28 | 22 |
| PagMTA1 | PagMTB1 | 2326.9 | −18.2 | 0.160 | 25 | 19 |
| PagMTA1 | PagMTB2 | 2321.8 | −15.9 | 0.265 | 25 | 13 |
| PagMTA1 | PagMTB3 | 2326.7 | −18.2 | 0.162 | 25 | 19 |
| PagMTA1 | PagMTB4 | 2321.8 | −15.9 | 0.265 | 25 | 13 |
| PagMTA2 | PagMTC1 | 2613.1 | −5.3 | 0.769 | 33 | 20 |
| PagMTA2 | PagMTC2 | 3360.1 | −14.0 | 0.325 | 28 | 18 |
| PagMTA2 | PagMTB1 | 2326.9 | −18.2 | 0.160 | 25 | 19 |
| PagMTA2 | PagMTB2 | 2321.8 | −15.9 | 0.265 | 25 | 13 |
| PagMTA2 | PagMTB3 | 2326.7 | −18.2 | 0.162 | 25 | 19 |
| PagMTA2 | PagMTB4 | 2321.8 | −15.9 | 0.265 | 25 | 13 |
| PagMTC1 | PagMTB2 | 2335.9 | −11.0 | 0.591 | 29 | 11 |
| PagMTC2 | PagMTB2 | 2391.4 | −7.8 | 0.442 | 25 | 18 |
| PagMTC1 | PagMTB1 | 2281.4 | −11.9 | 0.479 | 28 | 9 |
| PagMTC2 | PagMTB1 | 2445.5 | −8.6 | 0.398 | 25 | 22 |
| PagMTC1 | PagMTB3 | 2391.4 | −7.8 | 0.442 | 25 | 18 |
| PagMTC2 | PagMTB3 | 2281.1 | −11.9 | 0.481 | 28 | 9 |
| PagMTC1 | PagMTB4 | 2281.8 | −11.8 | 0.508 | 28 | 9 |
| PagMTC2 | PagMTB4 | 2391.4 | −7.8 | 0.442 | 25 | 18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Wu, W.; Yang, Y.; Wilson, I.; Shao, F.; Qiu, D. Genome-Wide Identification of m6A Writers, Erasers and Readers in Poplar 84K. Genes 2022, 13, 1018. https://doi.org/10.3390/genes13061018
Sun X, Wu W, Yang Y, Wilson I, Shao F, Qiu D. Genome-Wide Identification of m6A Writers, Erasers and Readers in Poplar 84K. Genes. 2022; 13(6):1018. https://doi.org/10.3390/genes13061018
Chicago/Turabian StyleSun, Xiaochen, Wenli Wu, Yanfang Yang, Iain Wilson, Fenjuan Shao, and Deyou Qiu. 2022. "Genome-Wide Identification of m6A Writers, Erasers and Readers in Poplar 84K" Genes 13, no. 6: 1018. https://doi.org/10.3390/genes13061018
APA StyleSun, X., Wu, W., Yang, Y., Wilson, I., Shao, F., & Qiu, D. (2022). Genome-Wide Identification of m6A Writers, Erasers and Readers in Poplar 84K. Genes, 13(6), 1018. https://doi.org/10.3390/genes13061018