1. Introduction
Segmental duplications (SD) are highly identical, 10–300 kb long genomic sequences present from two to a few times in the genome, either interspersed or in tandem [
1,
2,
3]. They predispose regions to copy number variants (CNVs) and may thus act as mutational hotspots [
4,
5,
6].
Titin (
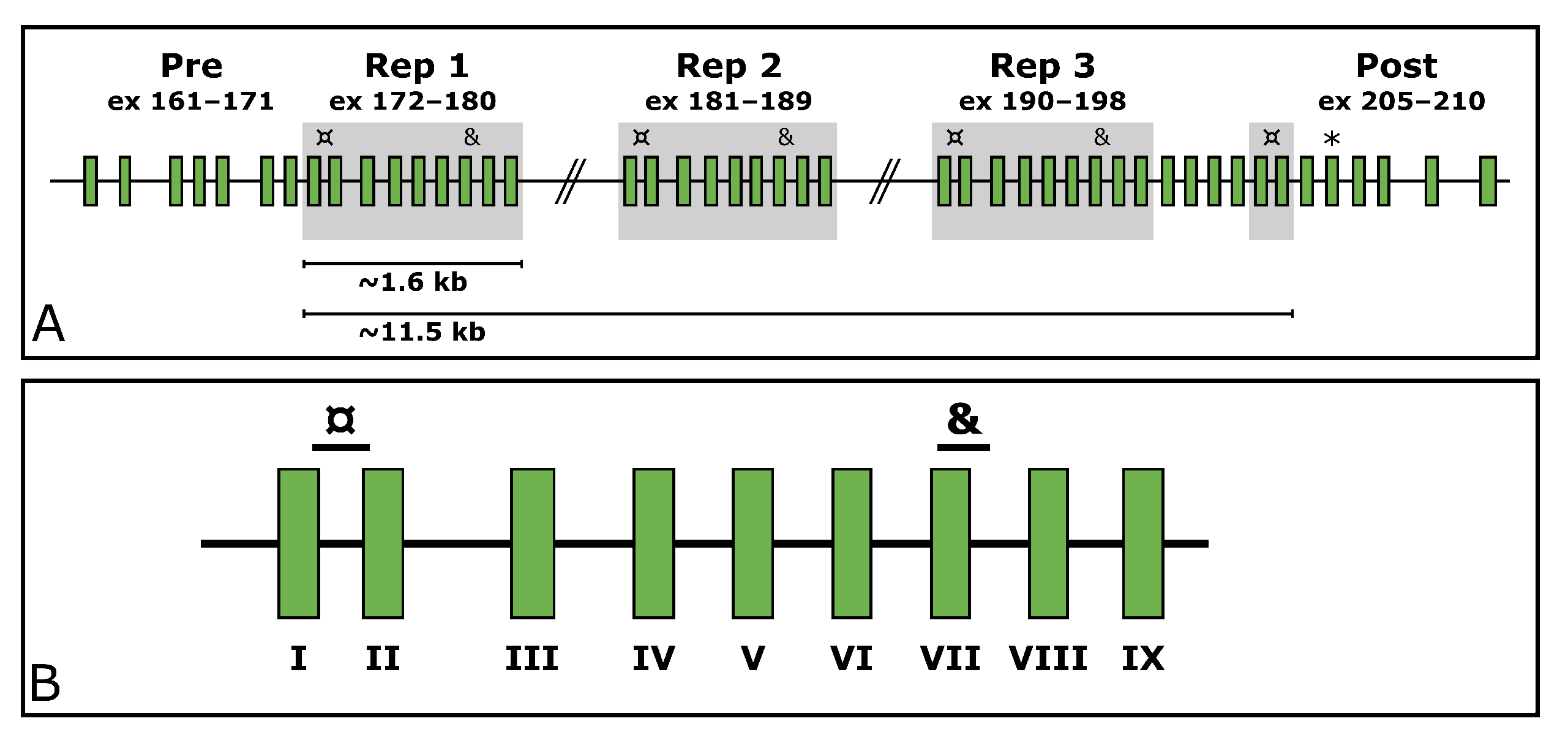
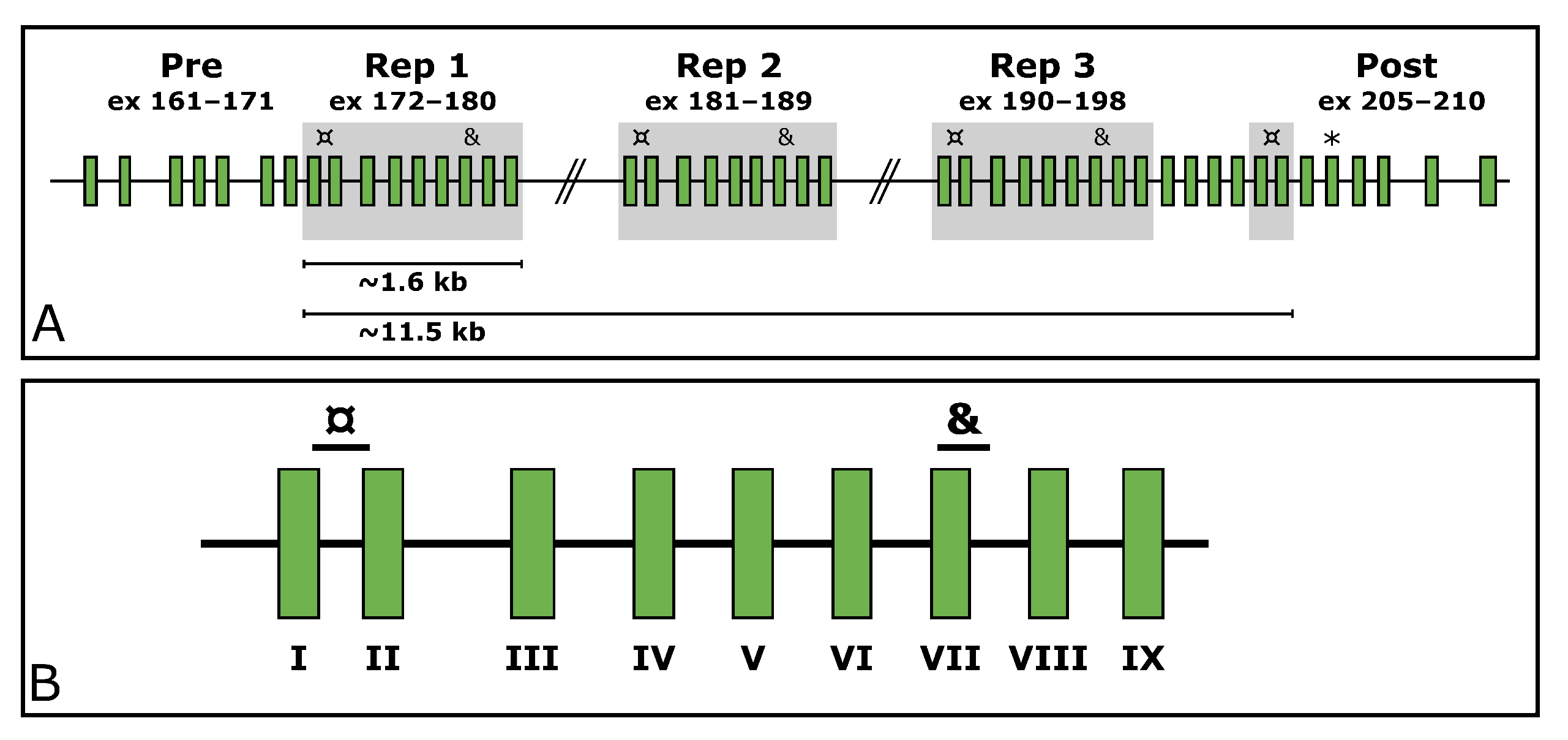
TTN, MIM ID *188840) is a gargantuan gene highly expressed in skeletal muscle. According to the reference sequence of the longest
TTN transcript (ENST00000589042.5, CCDS59435.1), it consists of 363 exons. In its middle, it holds a region encoding domains rich in proline (P), glutamate (E), valine (V), and lysine (K), referred to as the PEVK region [
7]. Within the PEVK region, it withholds a SD region (exons 172–180, 181–189, 190–198, and 203–204). This region consists of a 9-exon-block repeated three times, after which the two first exons of the block appear a fourth time. These two exons are separated by four exons from the last exon of the last repeated block (exon 198). The structure of the
TTN SD region is depicted in
Figure 1.
Mutations in
TTN can cause several different neuromuscular diseases, such as tibial muscular dystrophy and cardiomyopathies, in both recessive and dominant inheritance modes (MIM IDs dilated cardiomyopathy #604145, familial hypertrophic cardiomyopathy #613765, limb-girdle muscular dystrophy type 2J #608807, proximal myopathy #603689, Salih myopathy #611705, and tibial muscular dystrophy #600334) [
8,
9,
10,
11,
12].
The standard methods for routine CNV analysis are still microarray-based technologies, either using SNP or CNV probes, such as array Comparative Genomic Hybridisation (aCGH). However, CNV analysis methods based on massively parallel sequencing (MPS) data are rapidly improving in accuracy and reliability [
13,
14,
15,
16]. SDs and other repetitive regions still challenge both aCGH and methods based on MPS in CNV detection. Designing unique CNV or SNP probes is challenging due to the repetitive nature of these regions. Thus, these regions are typically avoided in commercial aCGH designs and often left with minimal to no probe coverage. Similarly, the alignment of short sequences challenges the analysis of repetitive regions with MPS-based methods.
We have previously designed and published two validated custom tiling CGH-arrays for the detection of CNVs in neuromuscular disorder genes [
17,
18]. These arrays also cover the
TTN SD and a similar SD in another muscle gene, the triplicate (TRI) region in nebulin (encoded by the
NEB gene, MIM ID *161650), to shed light on these regions and their variations [
17,
18].
Like
TTN,
NEB is a large structural protein highly expressed in skeletal muscle. Pathogenic variants in
NEB are a known cause of muscle disorders, such as nemaline myopathy (MIM #256030). Both
TTN and
NEB are thought to act as molecular templates, or rulers, for muscle filament length and structure [
19,
20,
21,
22,
23,
24]. As per this Ruler Hypothesis, large enough gains and losses in CN in the
NEB TRI and
TTN SD regions may be pathogenic [
19,
20,
21,
22,
23,
24,
25,
26]. It has been shown that gains of two blocks of
NEB TRI in one allele are, in fact, disease-causing [
25].
From a methodological molecular diagnostic perspective, the major difference between the SD regions of NEB and TTN is the difference in size—the NEB TRI covers altogether 30 kb of genomic region, which is roughly three times more than the TTN SD. Despite its repetitiveness, its length allows a tiled aCGH design in this region.
To allow for large-scale screening of CNVs of the
NEB TRI region, we previously developed two custom Droplet Digital PCR (ddPCR) assays targeting the region [
27]. Here, we present the extension of the study; custom ddPCR assays for the detection of CNVs within the
TTN SD region. The study aimed to use custom ddPCR assays to validate CNVs of the
TTN SD region in a cohort consisting of samples from neuromuscular disorder patients and family members previously studied using a custom CGH-array [
18]. Using ddPCR and aCGH data, we show that the
TTN SD region is subject to CNVs in a similar fashion to the
NEB TRI region. To our knowledge, this is the first publication acknowledging CNVs within
TTN SD to this degree and shows that this region should no longer be neglected in genetic analyses.
2. Materials and Methods
2.1. Samples
Altogether, 62 samples from 42 neuromuscular disorder families were acquired for the study. Of these, 42 were index patient samples, and the remaining 20 samples were from unaffected family members. The patient phenotypes included nemaline myopathy (n = 18), distal nemaline myopathy (n = 2), asymmetric distal myopathy (n = 1), cap myopathy (n = 1), unspecified congenital myopathy with arthrogryposis (n = 1), and unspecified congenital myopathy (n = 12). Nine of the patients had previously received a final molecular genetic diagnosis. Causative CNVs of the TTN SD region were not expected in the cohort.
The DNA stocks had been extracted either from peripheral blood or from saliva, eluted into EDTA, TE-buffer, or water, and stored at −20 °C. The DNA concentration and quality were checked with DeNovix DS-11 FX+ Spectrophotometer/Fluorometer (DeNovix Inc., Wilmington, DE, USA). Subsequent dilutions for the ddPCR reactions were performed in sterile water and stored at 4 °C.
2.2. Comparative Genomic Hybridisation Array Design, Protocol, and Analysis
All samples were run using a custom CGH-array for neuromuscular disorders (NMD-CGH-array) as previously described [
18].
The aCGH data were manually aligned for
TTN and
NEB to gain a zero baseline to avoid any subtle differences caused by the genome-wide normalisation of the analysis software (CytoSure Interpret Software v.4.11.30, Oxford Gene Technology Ltd, Cambridge, UK). The log
2 value for the
TTN SD region and large regions (124 kb and 157 kb) of the
TTN gene upstream and downstream of the SD were extracted from the aCGH data. The breakpoints used for normalisation of
TTN aCGH results were Chr2:(179389578_179390615)_(179512818_ 179513536) (upstream backbone), Chr2:(179533543_179533609)_(179691400_179690759) (downstream backbone), and Chr2:(179518163_179518846)_(179528302_179528492) (SD region). The breakpoints used for the normalisation of
NEB have been published earlier [
27].
The genomic locations for the aCGH data are given in the reference genome build Hg19/GRCh37. The normalised log2 value of the TTN SD region was acquired by subtracting the averaged background log2 value from the log2 value of the TTN SD region. The CNs of the SD region and the TTN backbone were estimated by converting the log2 values of the normalised TTN SD region and the average background log2 values to CNs assuming normal CNs of six and two, respectively.
2.3. Droplet Digital PCR
The ddPCR assays were designed, performed, and analysed according to the dMIQE guidelines [
28,
29]. The dMIQE checklist is available in
Supplemental Table S1.
2.3.1. Primer and Probe Design
Custom assays were designed for three regions in
TTN (NG_011618.3). Two of these target repeated exons within the SD region, and one targets an exon located downstream from the SD region (exon 206,
Figure 1A). The assays targeting the SD span from the end of exon 172/181/190/203 to the beginning of exon 173/182/191/204 (hereinafter referred to as
TTN SD exon I as per the
TTN SD exon it begins in) and from the beginning of exon 178/187/196 to the beginning of intron 178/187/196 (hereinafter referred to as
TTN SD exon VII) (
Figure 1B). The assay targeting the exon 206 downstream of the SD region is hereinafter referred to as
TTN Post-SD.
Primers and probes for the assays were designed using Primer3Plus (v.3.2.4,
https://www.primer3plus.com/index.html, accessed 17 May 2021) [
30,
31,
32]. Primers were designed to have a melting temperature (Tm) of approximately 60 °C, a GC content of 50–60%, a length of 20 bp, and avoiding putative secondary structures and G or C repeats over three bases long. The amplicons were not allowed to contain the BsuRI cut site sequence (GGCC) or the EcoRV cut site sequence (GATATC). Amplicon lengths vary from 101 to 120 bp.
Hydrolysis probes were designed to have a Tm of approximately 65 °C and a GC content of 30%–80%, aiming for a length of 25 bp. Custom probes were labelled with fluorescein amitide (FAM). Tm was calculated by the nearest neighbour method using OligoCalc (v.3.27,
http://biotools.nubic.northwestern.edu/OligoCalc.html, accessed 17 May 2021) [
33].
An in silico specificity screen was performed using the Standard Nucleotide BLAST blastn suite (v.2.11.0,
https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed 17 May 2021) [
34,
35], allowing four hits for the
TTN SD exon I assay primers and probes, three hits for the
TTN SD exon VII assay, and one hit for the
TTN Post-SD assay. The hits were confirmed to represent the respective targeted regions within
TTN.
All custom primer and probe sequences, along with amplicon lengths and locations within the reference sequences NG_011618.3 and NG_009382.2 are presented in
Supplemental Table S2. The reference used was a commercial
EIF2C1 ddPCR probe assay labelled with hexachloro-fluorescein (HEX) (cat. no. 10031243, Bio-Rad Laboratories Inc., Hercules, CA, USA).
EIF2C1, also known as Argonaute 1 (
AGO1), is a diploid gene located in the chromosomal region 1p34.3.
The previously published custom ddPCR assay for
NEB TRI exon VIII [
27] was included as a positive control for each sample in every run.
All assays were obtained from Bio-Rad Laboratories, Inc. (Hercules, CA, USA) as custom ordered primer/probe assays at a primer:probe ratio of 3.6:1. The concentrations were 900 nM of primer and 250 nM of probe in the final reaction mix.
2.3.2. Optimisation of ddPCR Assay Conditions
The optimal melting temperature for the assays was determined by running the reactions in a gradient PCR with different melting temperatures. The assays were evaluated using the melting temperatures of 57.5, 58.0, 58.5, 59.0, 59.5, 60.0, 60.5, and 61.0 °C. A melting temperature of 59.5 °C was chosen as it gave an adequate separation between the droplet clusters in all assays.
2.3.3. Assay Protocol
The total reaction volume was 20 μL, consisting of 10 ng of genomic template DNA in a volume of 7 μL, 1 μL of custom and reference assay each, 1 μL of restriction enzyme mix, and 10 μL of 2x ddPCR Supermix for Probes (No dUTPs) (cat. no. 1863023, Bio-Rad Laboratories Inc.). The restriction enzyme mix contained equal amounts of BsuRI (cat. no. FD0154, Thermo Scientific, Waltham, MA, USA) and EcoRV (cat. no. F0304, Thermo Scientific) for the TTN assays, and 1:1 diluted BsuRI in 10x FastDigest Buffer (cat. no. B64, Thermo Scientific) for the NEB assay. For each reaction, 22 μL of reaction mix was prepared, of which 20 μL was pipetted onto the DG8 Cartridge (cat. no. 1864008, Bio-Rad Laboratories Inc.). The cartridges were covered with DG8 Gaskets (cat. no. 1863009, Bio-Rad Laboratories Inc.).
The reactions were divided into approximately 1 nl droplets using the Droplet Generator QX2000 (Bio-Rad Laboratories Inc.) with Droplet Generator Oil (cat. no. 1863005, Bio-Rad Laboratories Inc.), transferred to ddPCR 96-well plates (cat. no. 12001925, Bio-Rad Laboratories Inc.) by pipetting and sealed with the PX1 PCR Plate Sealer (Bio-Rad Laboratories Inc.). The PCR reaction was performed using the DNA Engine Tetrad 2 Thermal Cycler (Bio-Rad Laboratories Inc.). The cycling steps were 95 °C 10 min; 40 cycles of (94 °C 30 s, 59.5 °C 1 min); 98 °C 10 min; 4 °C hold, with a ramp rate of 2 °C/s. The data were then visually inspected on the QuantaSoft Analysis v. 1.7.4.0917 and QuantaSoft Analysis Pro v. 1.0.596.0525 (Bio-Rad Laboratories Inc.) programs.
Each plate contained at least one no-template control for each assay to assess for putative contamination. All samples were run in duplicate.
2.3.4. Data Extraction and Filtering
Droplet data were extracted as comma-separated values (CSV) files from the QuantaSoft Analysis software and imported into Microsoft Excel.
Subsequent filtering was performed in Microsoft Excel using a minimum threshold of 25 droplets for individual droplet categories and 8500 accepted droplets per reaction. Data from two successful wells were used in subsequent analyses. In cases of more than two successful wells for the same assay and sample, the two runs with the largest number of accepted droplets were used in the analysis. The data were grouped as normal, gain, and loss samples by the predicted TTN SD copy number category based on the respective NMD-CGH-array results.
The range, mean, standard deviation (), and coefficient of variation in percent (%CV) were extracted and calculated of the accepted droplet number and target copies per 20 μL, and for the estimated CN by ddPCR and aCGH for the respective regions targeted by the ddPCR assays.
2.4. Statistical Analysis
To assess the performance of the ddPCR assays in CN classification, we performed one-way analysis of variance (ANOVA) tests and post hoc Tukey’s honest significant difference (Tukey’s HSD) test using the ddPCR derived CN and the aCGH-predicted CNV class (either normal, loss, or gain). Additionally, we used Bland–Altman analysis [
35] to assess the agreement between the method using the ddPCR derived CN and the aCGH-estimated CN.
A linear regression analysis was performed using the CN estimates from ddPCR and aCGH. In addition, the Pearson correlation coefficient was also calculated from these data. A Pearson correlation coefficient of >0.70 was considered a strong correlation.
To assess reproducibility within experiments, we performed intra-assay analyses separately for all assays using duplicates run within the same experiment. The analysis included calculations of %CV and
of the differences between repeated measurements [
35].
To assess repeatability between experiments, we performed inter-assay analyses separately for all assays using duplicates run on separate plates. The analysis included calculations of %CV and
of the differences between repeated measurements [
35].
The accuracy, sensitivity, and specificity of the assays were assessed as described in [
36], by grouping the samples by their aCGH estimated CN for respective targeted region. For the
TTN SD exon I assay, ddPCR CN values within the interval [7.5, 8.5] were considered normal. For the
TTN SD exon VII and
NEB TRI exon VIII assays, ddPCR CN values within the interval [5.5, 6.5] were considered normal. For the
TTN Post-SD assay, ddPCR CN values between [1.5, 2.5] were considered normal. Values subceeding and exceeding the given intervals were classified as losses and gains as per the ddPCR results, respectively.
All statistical analyses were performed in RStudio (v.1.4.1103) using R v.4.0.4. [
37,
38]. Boxplots were generated using ggplot2 v.3.3.6 [
39].
3. Results
3.1. Data Overview
Altogether 55 samples passed the initial quality filtering in all assays in at least two parallel wells. Of these, 36 were samples from neuromuscular disorder patients and 19 were samples from unaffected family members. Of all samples, 35 were classed as normal (CN = 6), 11 as losses (CN < 6), and 9 as gains (CN > 6) as per the CNV prediction of the NMD-CGH-array for the TTN SD region. Gains and losses in the TTN SD region were present in both patient and unaffected family member samples.
Table 1 shows the number of samples representing different predicted CN classes of the
TTN SD and the CN distribution between affected and unaffected individuals. The expected CN of the
TTN SD region as per the NMD-CGH-array data varied between 5 and 11 in the samples analysed. The expected CN of the
NEB TRI region varied between 4 and 11, and an average CN of 6 was classified as normal for the
NEB TRI also.
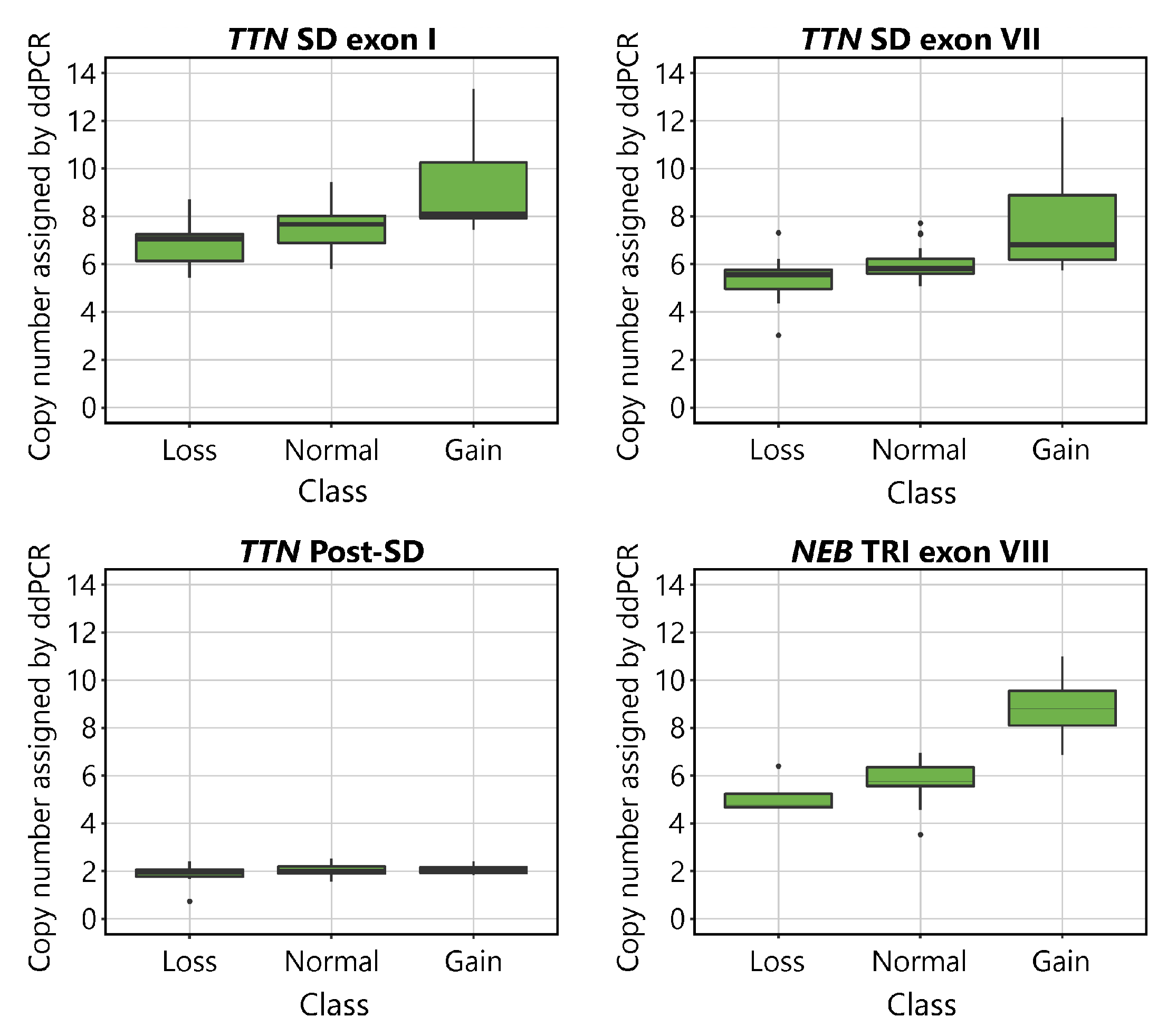
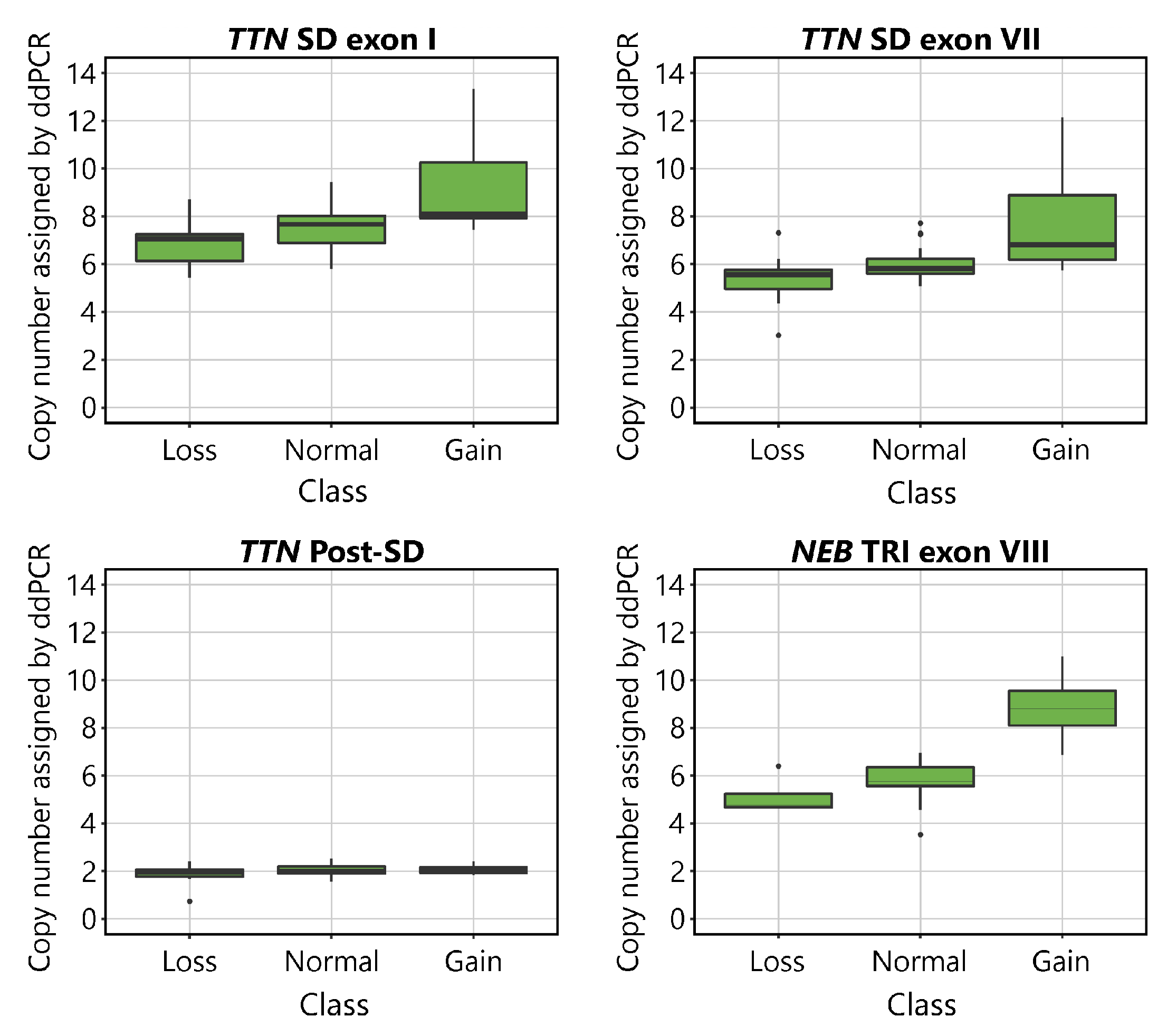
The CNs indicated by ddPCR are shown plotted against the aCGH-determined CN class for the
TTN SD and
NEB TRI in
Figure 2. Visual inspection of the plots suggests that the assays detect differences between samples belonging to different CN groups. The mean values of the normal groups approach the expected values of 8 for the
TTN SD exon I, 6 for
TTN SD exon VII and
NEB TRI exon VIII, and 2 for
TTN Post-SD. As expected, differences were seen between groups in all assays except for the
TTN Post-SD assay.
The mean accepted droplet count was 15,644 (
= 2159.9, %CV = 13.8) for the
TTN SD exon I assay, 15,501 (
= 2322.3, %CV = 15.0) the
TTN SD exon VII assay, 15,580 (
= 2482.4, %CV = 15.9) for the
TTN Post-SD assay, and 15,042 (
= 2122.0, %CV = 14.1) for the
NEB TRI exon VIII assay. The range, mean,
, and %CV values for accepted droplet numbers, target, and reference copies per 20 μL are presented in
Supplemental Table S3 broken down by the assigned CN groups. Droplet numbers showed no significant differences between assays or groups. The means of the target copies per 20 μL were distributed as expected, with the highest number of target copies in the
TTN SD exon I assay, equal and lower number of target copies in the
TTN SD exon VII and
NEB TRI exon VIII assays, and the lowest number of target copies in the
TTN Post-SD assay. The range, mean,
, and %CV values for the ddPCR and aCGH estimated CNs for each ddPCR targeted sequence are presented in
Supplemental Table S4.
The difference between the CNs estimated by the TTN SD exon I and TTN SD exon VII assays was normally distributed around the mean value of 1.61, [, 3.51]. The mean difference was marginally larger in the normal group (1.63, [−0.85, 3.51]) as compared with the gain (1.59, [1.11, 2.43]) and loss (1.55, [0.92, 2.50]) groups. In the normal group, the expected difference was 2.00, to account for the number of times the amplicon sequences are expected to occur in the genome.
3.2. One-Way ANOVA and Tukey’s HSD
The one-way ANOVA indicated statistically significant differences between the ddPCR results for at least two different groups in assays TTN SD exon I (F(2,52) = [9.80], p < 0.001), TTN SD exon VII (F(2,52) = [11.89], p < 0.00001), and NEB TRI exon VIII (F(2,52) = [32.95], p < 0.00001). No statistically significant differences were found between the groups in the TTN Post-SD assay (F(2,52) = [1.83], p = 0.171), as expected.
Tukey’s HSD test for multiple comparisons found that the mean value of the ddPCR results was significantly different between the normal and gain, and loss and gain groups in all assays except for the TTN Post-SD assay. No statistical significance was found between the normal and loss groups in any assays.
3.3. Pearson Correlation
All Pearson correlation coefficients, except that for the
TTN Post-SD assay against its corresponding
TTN SD aCGH CN, exceeded the set threshold of 0.70 for a strong correlation. The Pearson correlation coefficients are presented in
Table 2.
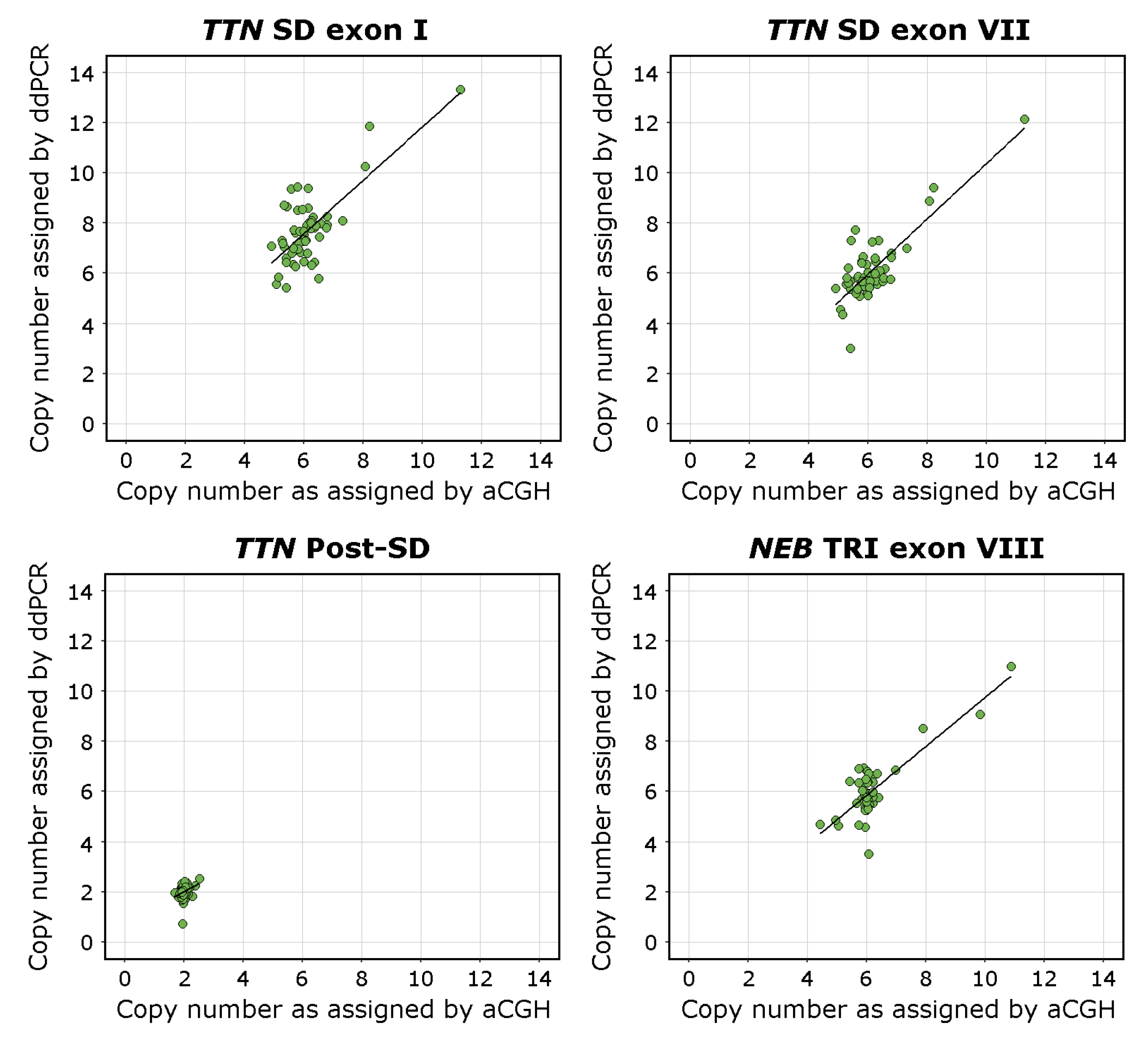
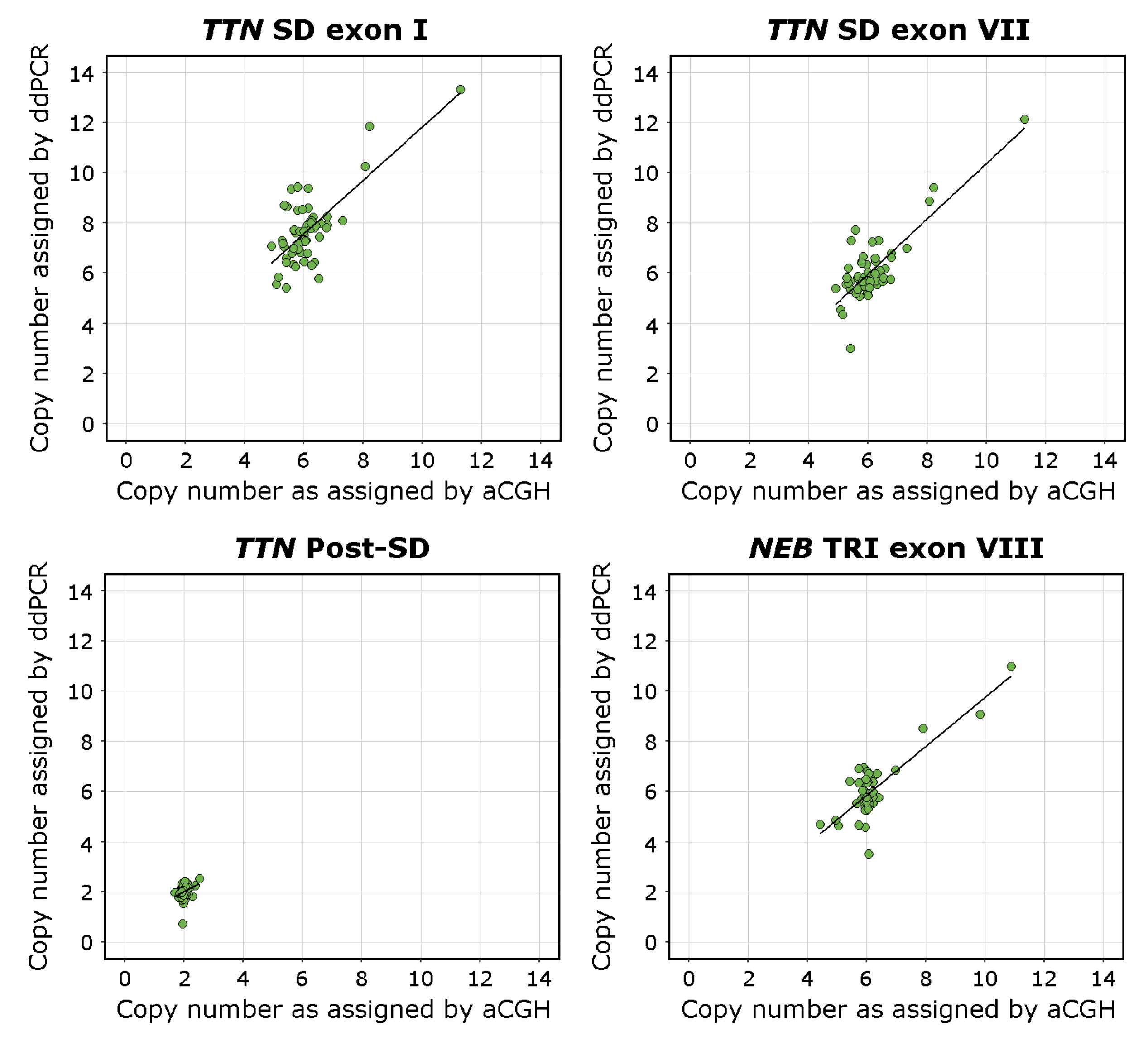
3.4. Linear Regression
The overall regression was statistically significant for the
TTN SD exon I assay (adjusted R
2 = 0.53, F(1,53) = 50.59,
p < 0.00001), the
TTN exon VII assay (adjusted R
2 = 0.66, F(1,53) = 104.9,
p < 0.00001), and the
NEB TRI exon VIII assay (adjusted R
2 = 0.67, F(1,53) = 113,
p < 0.00001). The linear regression analysis found that the ddPCR assays seemed to recognise CNVs of the
TTN SD region adequately. The
TTN SD exon VII assay approached the already validated
NEB TRI exon VIII assay [
27] in its accuracy.
The overall regression of the TTN Post-SD assay CN estimates against the aCGH estimated CN of the TTN backbone (excluding the SD) was statistically significant (adjusted R2 = 0.07, F(1,53) = 5.1, p < 0.05). The adjusted R2 value approaching 0 indicates that the sequence targeted by the TTN Post-SD assay indeed lies outside the TTN SD region and that it adequately amplifies a diploid region. The overall regression of the TTN Post-SD assay CN estimates against the aCGH estimated CN of the TTN SD region was not statistically significant (adjusted R2 = −0.002, F(1,53) = 0.89, p = 0.351), which further supports this conclusion.
The overall regression of the TTN SD exon I assay CN estimates against the TTN SD exon VII assay CN estimates was statistically significant (adjusted R2 = 0.75, F(1,53) = 160.5, p < 0.00001), indicating concordance between the assays.
The complete results of the linear regression models are presented in
Supplemental Table S6. Visualisations of the linear regression analyses and scatterplots of the data are presented in
Figure 3.
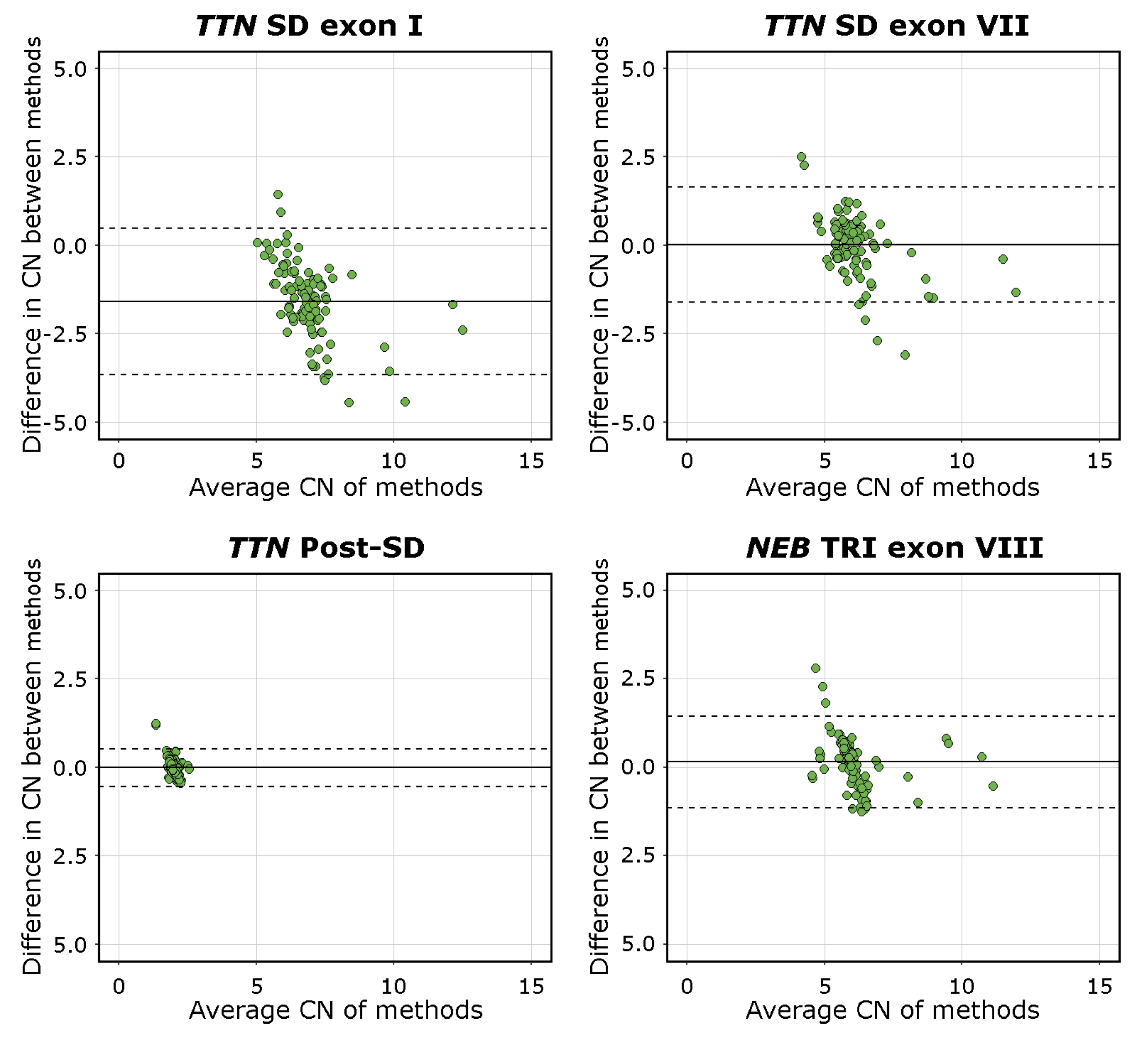
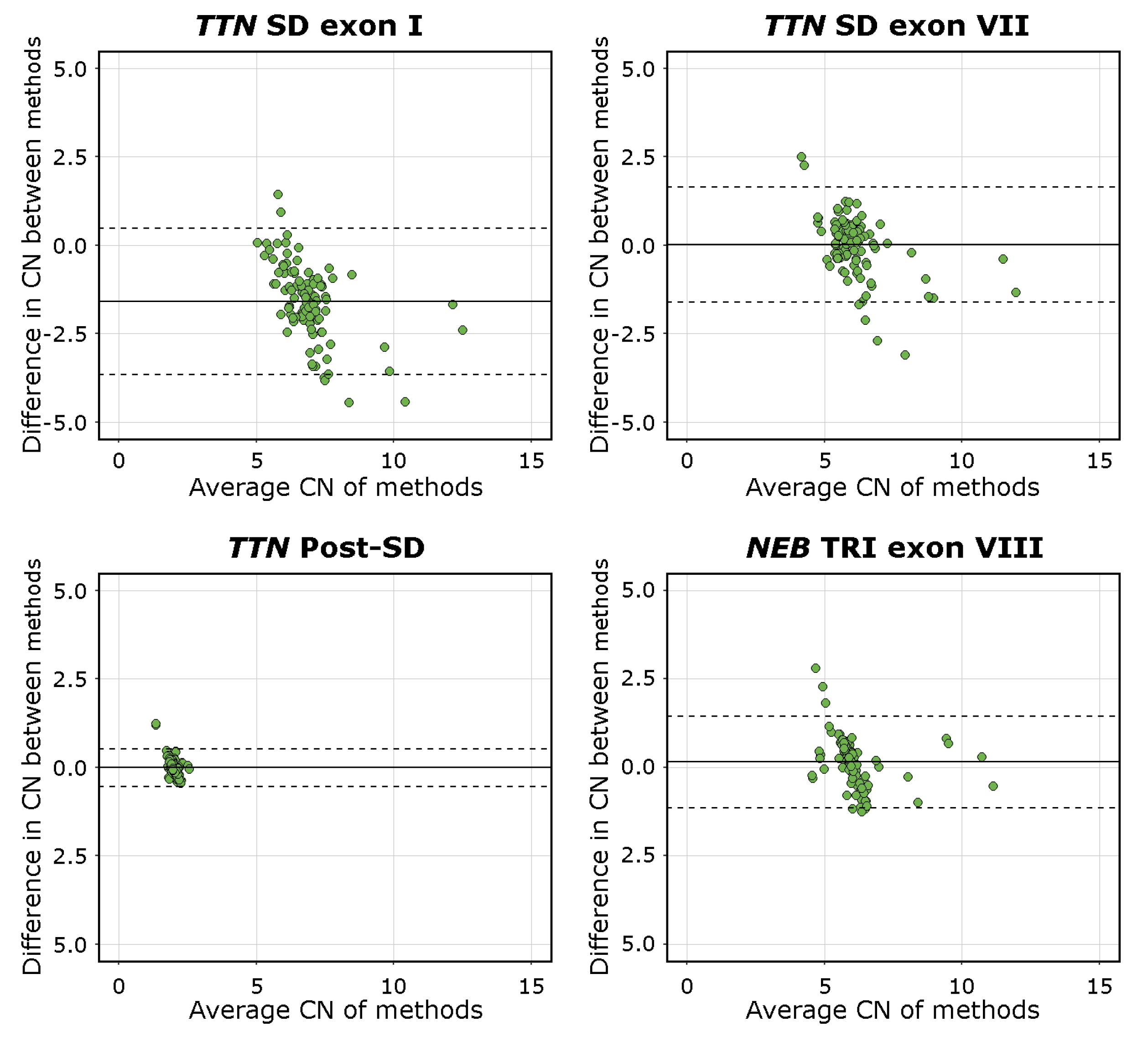
3.5. Bland–Altman Analysis
The mean difference was −1.59 [95%C.I. −3.65, 0.48] for
TTN SD exon I, 0.02 [95%C.I. −1.61, 1.65] for
TTN SD exon VII, −0.01 [95%C.I. −0.54, 0.52] for
TTN Post-SD, and 0.13 [95%C.I. −1.15, 1.45] for
NEB TRI exon VIII. The visualisations of the Bland–Altman analysis are shown in
Figure 4. Mean values approaching 0 indicate higher levels of agreement between the methods compared. The
TTN SD exon VII,
TTN Post-SD, and
NEB TRI exon VIII assays, therefore, performed adequately, while the mean difference and scatter in differences in the
TTN SD exon I results indicate a lower degree of agreement between the methods. This is consistent with the fact that the
TTN SD exon I and exon II CN is higher than the average CN of the entire repeated block, which the NMD-CGH array cannot detect due to methodological limitations. The complete Bland–Altman analysis data, including 95%C.I. for the limits of agreement, are presented in
Supplemental Table S7.
3.6. Intra-Assay and Inter-Assay Analyses
To assess the reproducibility of the assays, the intra-assay duplicate means, , and differences, and %CV were calculated along with mean %CV, mean difference () and mean of differences () for all samples per assay. To assess the repeatability of the assays, the inter-assay duplicate means, , differences, and %CV were calculated along with mean %CV, , and of differences for all samples per assay.
The summarised intra-assay and inter-assay analysis results are presented in
Table 3. The complete intra-assay and inter-assay analyses with underlying data are presented in
Supplemental Tables S8 and S9, respectively.
3.7. Accuracy, Sensitivity, and Specificity
The TTN SD exon I assay yielded an accuracy of 0.218, a sensitivity of 0.667, and a specificity of 0.389. The TTN SD exon VII assay yielded an accuracy of 0.582, a sensitivity of 0.526, and a specificity of 0.639. The TTN Post-SD assay yielded an accuracy of 0.964 and a specificity of 0.964. Sensitivity for the TTN Post-SD assay could not be calculated due to the lack of both true positives and false negatives when assessed against the TTN backbone. The NEB TRI exon VIII assay yielded an accuracy of 0.691, a sensitivity of 0.875, and a specificity of 0.660.
4. Discussion
Our novel ddPCR assays confirm the presence of recurring CNVs within the
TTN SD region, as first seen on the NMD-CGH-array [
18]. CNVs in the
TTN SD region are about three times more common than CNVs in the
NEB TRI region. To our knowledge, this is the first time that CNVs within this region are acknowledged to this degree.
Our custom ddPCR assays for TTN SD exon I and TTN SD exon VII reliably recognise samples with gains in the TTN SD region, as supported by the statistical tests performed.
These gains were also detected by the NMD-CGH-array [
18]. However, custom CGH-array methodology is challenged by the sequence length of the
TTN SD region. Furthermore, the
TTN SD is doubly repetitive; on top of the block repeating, the exons within the block are similar and share sequences with flanking exons. Therefore, the resolution and mathematical mode of analysis of aCGH data are challenged by this region, as it heavily relies on the possibility of incorporating enough unique and tiled probes, and aberration calls are typically made based on several consecutive affected probes.
There was no statistical significance between the normal and loss groups in the
TTN SD exon I and
TTN SD exon VII assays. This is presumably due to two factors: the relatively low number of samples and thus the lack of power in the statistical methods and the lack of CN amplitude variance within the deletion group. We have yet to see losses of two or more copies of the
NEB TRI region in the over 430 samples that we have previously analysed on our custom NM- and NMD-CGH-arrays [
17,
18]. We have, however, seen cases with up to eight extra copies, yielding a total CN of 14 of the
NEB TRI. It is also possible that at least part of the suspected
TTN SD region losses are methodological artefacts due to the relatively low number of good quality aCGH probes that can be designed in the region. This, in turn, is limited by the repetitiveness and length of the
TTN SD region.
The ddPCR method for detecting CNVs within the TTN SD could theoretically be improved by designing more assays covering other sequences of the region. In our experience, however, in silico optimisation does not necessarily guarantee working assays. Furthermore, the exons within the repeating block of the TTN SD are highly similar, limiting the possibilities for unique assay design.
Our study was also affected by the samples’ heterogeneous background in terms of DNA extraction method and storage; the method would most probably significantly benefit from streamlining the DNA extraction process. Different DNA extraction methods, the quality of the DNA, and possible amplification or fluorescent chemistry inhibitors may well affect the ddPCR assay results. It is difficult to conclude which factors are marginally influential and which affect the end result significantly with a variable sample collection. Our previous study observed that diversity in DNA extraction methods and quality of DNA does introduce a level of variability in the results [
27].
The intra- and inter-assay analyses showed adequate reproducibility and acceptable repeatability. The larger repeatability %CV values may be explained by differences in manual handling, such as pipetting techniques between the persons who produced the data. Both intra- and inter-assay analysis results could improve by using automated pipetting machinery.
The
TTN SD is part of the PEVK-encoding region of titin. This region undergoes extensive tissue-specific alternative splicing giving rise to cardiac and skeletal muscle-specific isoforms. The
TTN SD exons seem to be missing from the cardiac isoforms [
40]. The PEVK region forms an elastic spring that modulates titin-based force in skeletal muscle through interactions with calcium and actin [
41].
What remains to be elucidated is the exact CN of the TTN SD gains and losses, along with the detailed structure and orientation of gains. Furthermore, phenotype-genotype correlation analyses are needed to pinpoint a pathogenic threshold of the TTN CNVs. It is unlikely that a single repeat block gain or loss would significantly alter the phenotype, seeing the size of the gene and its product. However, we hypothesise that a large enough size deviation of the PEVK region from normal may affect the elastic properties of titin and force generation in skeletal muscle.
Approximately 5% of the human genome consists of SD regions, and they are a significant feature of mammalian genomes [
1,
2,
3,
4]. Repetitive regions, including SDs, are often neglected in both MPS-based and aCGH methods. While sequencing technology and analysis of sequence data have come a long way in terms of CNV detection [
14,
15], the methodology has significant limitations regarding repetitive sequences. DNA amplification is a common step in MPS protocols, and in the case of repeated regions, it may severely distort the actual repeat number in the subsequent analysis. More importantly, the length of the contigs that modern MPS technologies produce is insufficient to get a reliable alignment of these repetitive regions, which may span several megabases.
Attempts to sequence over the NEB TRI and TTN SD regions have been unsuccessful, as even the long-read sequencing methods often rely on an amplification step. As long-read sequencing methods develop in due time, we hope to sequence over both the NEB TRI and TTN SD regions.




{kind=link}
{kind=link}
{kind=link}
{kind=link}