
Comparative Genomic Hybridization to Microarrays in Fetuses with High-Risk Prenatal Indications: Polish Experience with 7400 Pregnancies

 , , , , , , , , , , , , ,
, , , , , , , , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
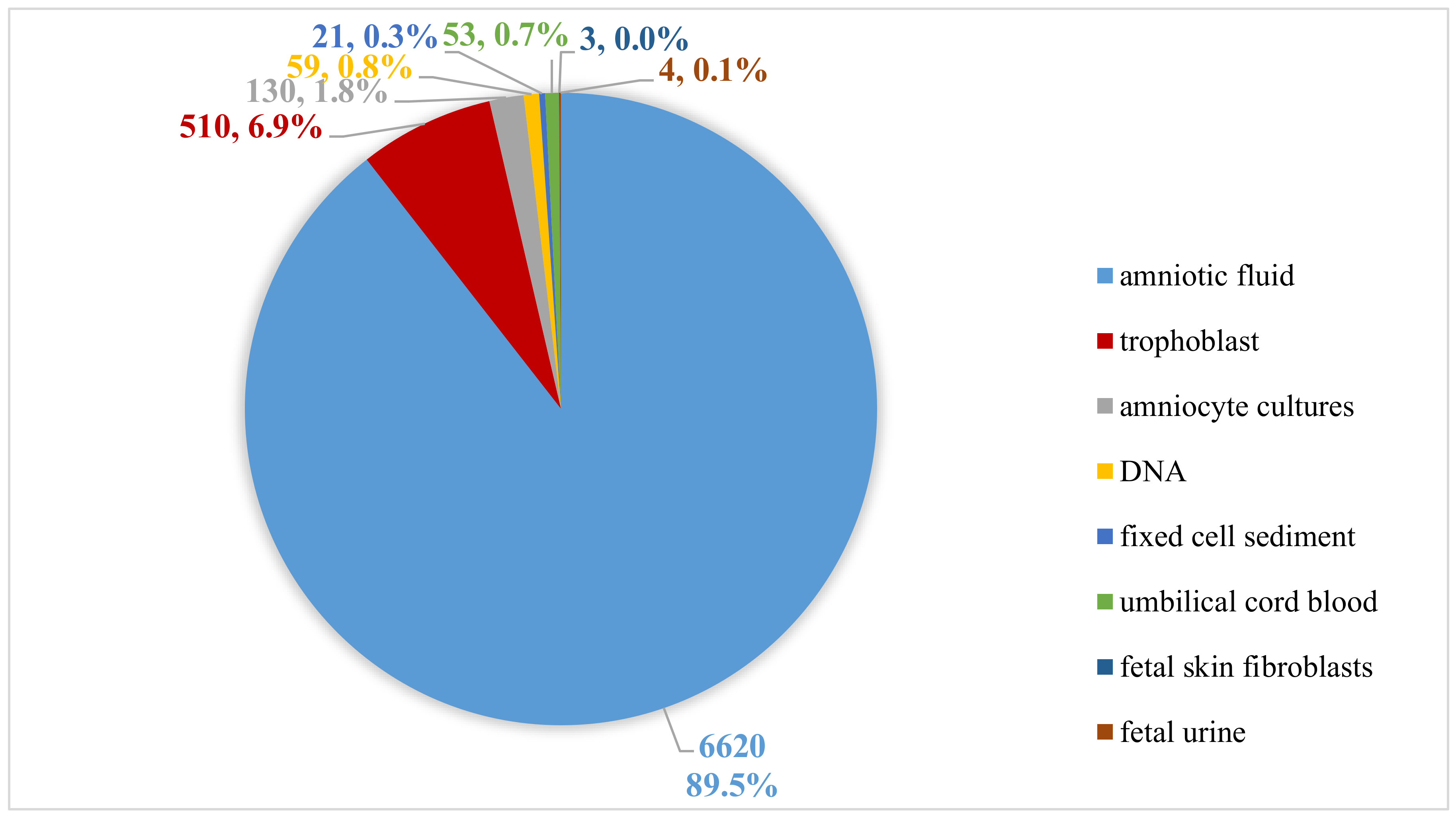
2.1. Sample Types and DNA Isolation
2.2. Genomic Array Platform (Array Comparative Genomic Hybridization (Array CGH) Analysis and Interpretation)
2.3. CNV Classification
- Pathogenic aberrations: CNV was classified as pathogenic if it was a large aberration of several MB, or was associated with known microdeletion/microduplication syndromes or contained known genes responsible for a specific pathology and had been previously described in specific clinical disorders.
- Likely pathogenic aberrations: CNVs that have not yet been described or have been rarely described and contain some gene/genes whose function is known and most likely may be responsible for the clinical features of the patient.
- Variants of unknown significance (VOUS): This category includes CNVs that did not have a clearly defined clinical significance at the time of publication of the study results. These changes were not included in the prenatal outcomes because the function of the genes in this region was unknown or the cause of the abnormal ultrasound examination was difficult to determine. In our study, parental inheritance of VOUS was not specified.
- Likely benign aberrations: CNVs that occur in healthy people and have only been described in a few cases in the general population, but do not represent a common polymorphism. CNV interpreted as possibly mild was not reported in a result of the study.
- Benign aberrations: do not affect the phenotype (polymorphisms occurring in the general population), which include: aberrations in the region of segmental duplication, aberrations that do not contain genes, aberrations in regions containing dose-insensitive genes frequently repeated in the Polish population and known aberrations as copy number variants described in the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home (accessed on 3 January 2022)) (path: DGV Gold Standard Variants).
3. Results
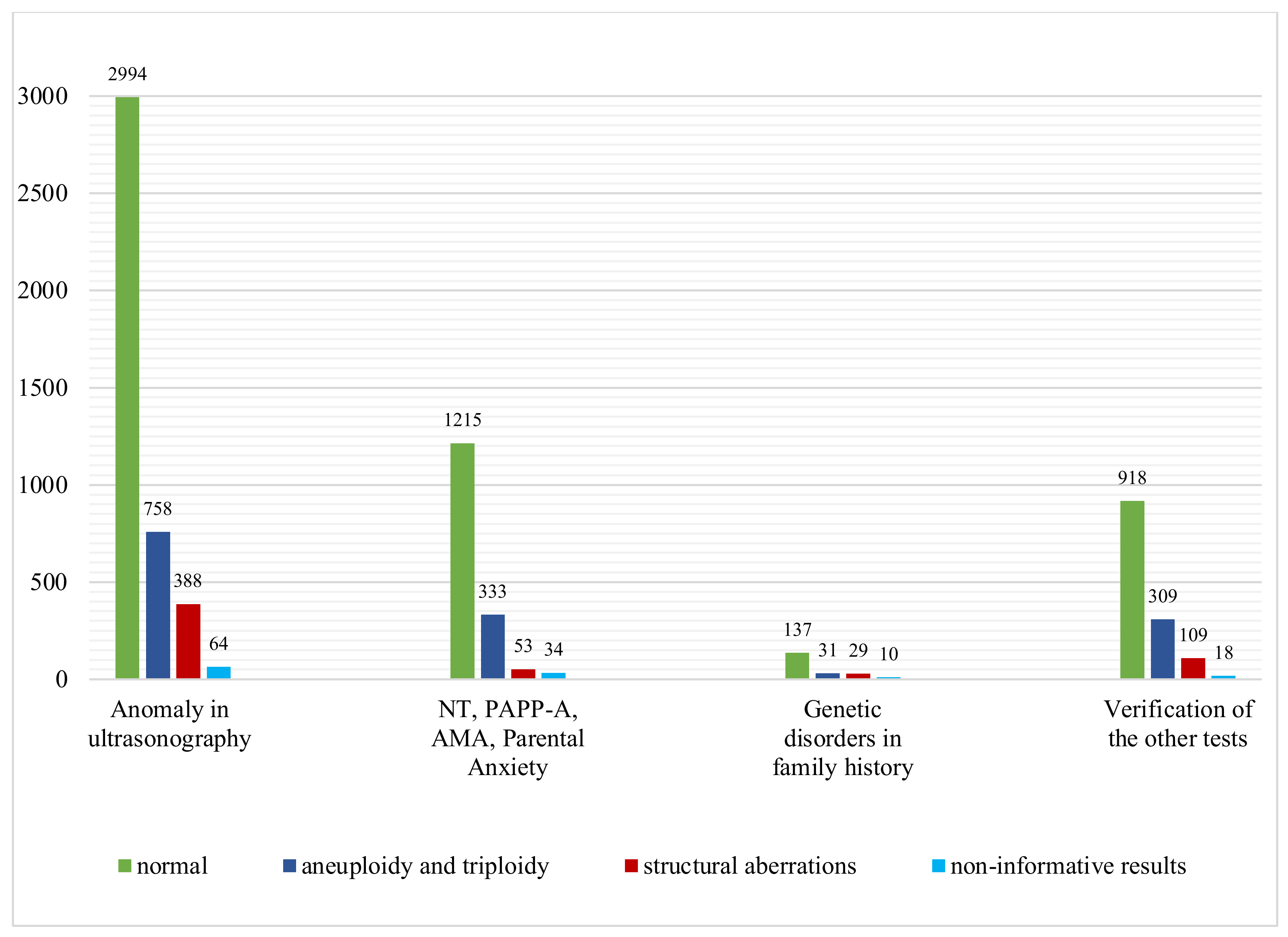
3.1. Anomaly on Ultrasonography
3.2. Advanced Maternal Age (AMA)
3.3. Abnormal Serum Screening Results: PAPP-A
3.4. Other Indications
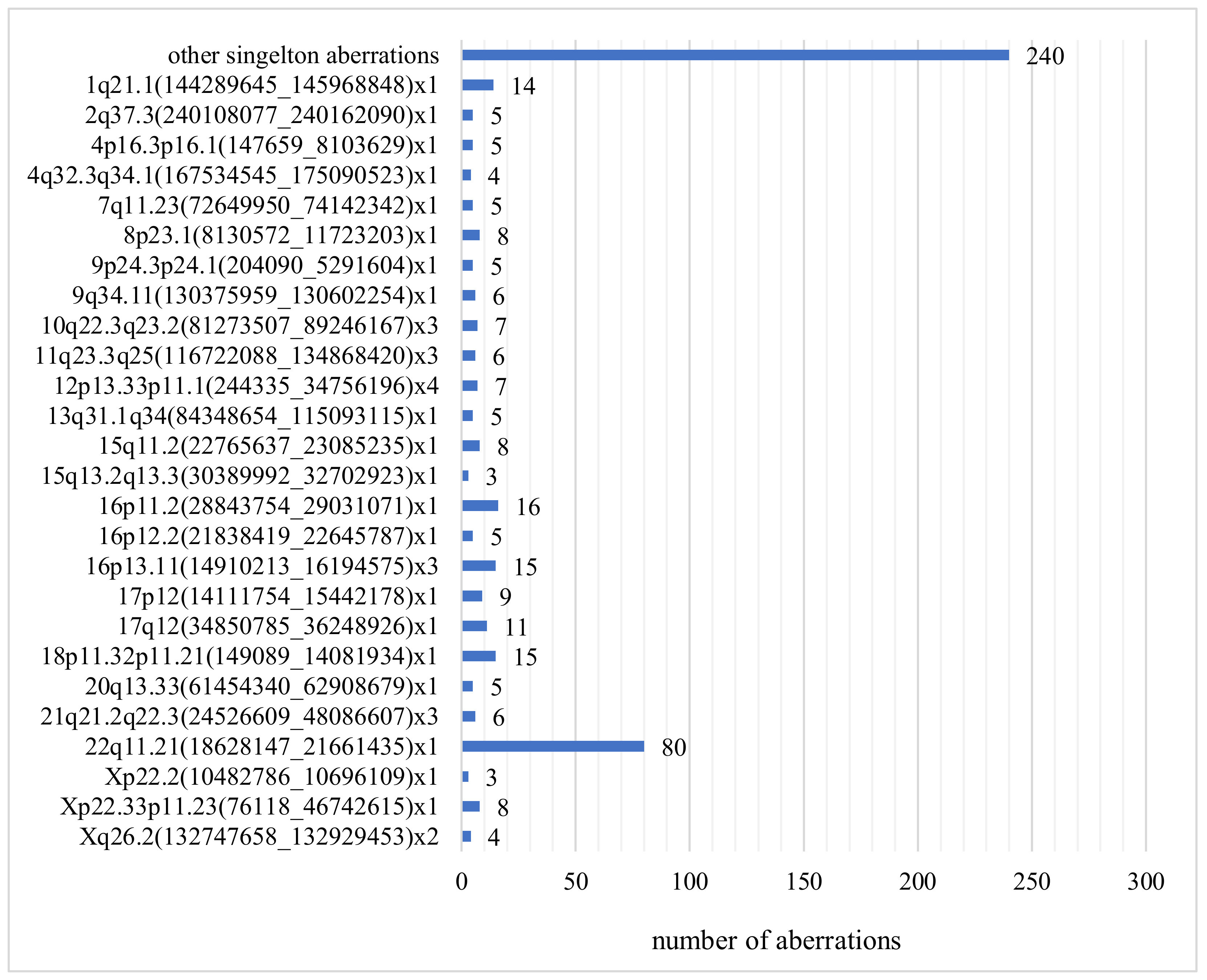
3.5. Discrepancy between Karyotyping and CGH Array
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luo, X.; Zhu, H.; Wang, L.; Xiao, B.; Fan, Y.; Ye, H.; Ying, X.; Qiu, W.; Zhang, H.; Han, L.; et al. Chromosomal microarray analysis in fetuses with high-risk prenatal indications: A retrospective study in China. Taiwan. J. Obstet. Gynecol. 2021, 60, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Ding, Y.; Song, X.; Mao, J.; Liu, M.; Liu, Y.; Huang, C.; Zhang, Q.; Wang, T. Clinical Utility of SNP Array Analysis in Prenatal Diagnosis: A Cohort Study of 5000 Pregnancies. Front. Genet. 2020, 11, 571219. [Google Scholar] [CrossRef] [PubMed]
- Robson, S.C.; Chitty, L.S.; Morris, S.; Verhoef, T.; Ambler, G.; Wellesley, D.G.; Graham, R.; Leader, C.; Fisher, J.; Crolla, J.A. Evaluation of Array Comparative Genomic Hybridisation in prenatal diagnosis of fetal anomalies: A multicentre cohort study with cost analysis and assessment of patient, health professional and commissioner preferences for array comparative genomic hybridisation. Effic. Mech. Eval. 2017, 4, 1–104. [Google Scholar]
- Stosic, M.; Levy, B.; Wapner, R. The Use of Chromosomal Microarray Analysis in Prenatal Diagnosis. Obstet. Gynecol. Clin. N. Am. 2018, 45, 55–68. [Google Scholar] [CrossRef]
- Mademont-Soler, I.; Morales, C.; Soler, A.; Martínez-Crespo, J.M.; Shen, Y.; Margarit, E.; Clusellas, N.; Obón, M.; Wu, B.L.; Sánchez, A. Prenatal diagnosis of chromosomal abnormalities in fetuses with abnormal cardiac ultrasound findings: Evaluation of chromosomal microarray-based analysis. Ultrasound Obstet. Gynecol. 2013, 41, 375–382. [Google Scholar] [CrossRef]
- Committee on Genetics and the Society for Maternal-Fetal Medicine. Committee Opinion No. 682: Microarrays and Next-Generation Sequencing Technology: The Use of Advanced Genetic Diagnostic Tools in Obstetrics and Gynecology. Obstet. Gynecol. 2016, 128, e262–e268. [Google Scholar] [CrossRef]
- Hay, S.B.; Sahoo, T.; Travis, M.K.; Hovanes, K.; Dzidic, N.; Doherty, C.; Strecker, M.N. ACOG and SMFM guidelines for prenatal diagnosis: Is karyotyping really sufficient? Prenat. Diagn. 2018, 38, 184–189. [Google Scholar] [CrossRef]
- Armour, C.M.; Dougan, S.D.; Brock, J.A.; Chari, R.; Chodirker, B.N.; DeBie, I.; Evans, J.A.; Gibson, W.T.; Kolomietz, E.; Nelson, T.N.; et al. Practice guideline: Joint CCMG-SOGC recommendations for the use of chromosomal microarray analysis for prenatal diagnosis and assessment of fetal loss in Canada. J. Med. Genet. 2018, 55, 215–221. [Google Scholar] [CrossRef]
- Silva, M.; de Leeuw, N.; Mann, K.; Schuring-Blom, H.; Morgan, S.; Giardino, D.; Rack, K.; Hastings, R. European guidelines for constitutional cytogenomic analysis. Eur. J. Hum. Genet. 2019, 27, 1–16. [Google Scholar] [CrossRef]
- Song, T.; Wan, S.; Li, Y.; Xu, Y.; Dang, Y.; Zheng, Y.; Li, C.; Zheng, J.; Chen, B.; Zhang, J. Detection of copy number variants using chromosomal microarray analysis for the prenatal diagnosis of congenital heart defects with normal karyotype. J. Clin. Lab. Anal. 2019, 33, e22630. [Google Scholar] [CrossRef]
- Lee, M.Y.; Won, H.S.; Han, Y.J.; Ryu, H.M.; Lee, D.E.; Jeong, B.D. Clinical value of chromosomal microarray analysis in prenatally diagnosed dextro-transposition of the great arteries. J. Matern. Fetal Neonatal Med. 2020, 33, 1480–1485. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.L.; Li, R.; Fu, F.; Pan, M.; Han, J.; Yang, X.; Zhang, Y.L.; Li, F.T.; Liao, C. Chromosome microarray analysis in the investigation of children with congenital heart disease. BMC Pediatr. 2017, 17, 117. [Google Scholar] [CrossRef] [PubMed]
- Ciaccio, C.; Fontana, L.; Milani, D.; Tabano, S.; Miozzo, M.; Esposito, S. Fragile X syndrome: A review of clinical and molecular diagnoses. Ital. J. Pediatr. 2017, 43, 39. [Google Scholar] [CrossRef]
- Wapner, R.J.; Martin, C.L.; Levy, B.; Ballif, B.C.; Eng, C.M.; Zachary, J.M.; Savage, M.; Platt, L.D.; Saltzman, D.; Grobman, W.A.; et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N. Engl. J. Med. 2012, 367, 2175–2184. [Google Scholar] [CrossRef] [PubMed]
- Srebniak, M.I.; Diderich, K.E.; Joosten, M.; Govaerts, L.C.; Knijnenburg, J.; de Vries, F.A.; Boter, M.; Lont, D.; Knapen, M.F.; de Wit, M.C.; et al. Prenatal SNP array testing in 1000 fetuses with ultrasound anomalies: Causative, unexpected and susceptibility CNVs. Eur. J. Hum. Genet. 2016, 24, 645–651. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Rosenfeld, J.A.; Dabell, M.P.; Coppinger, J.; Bandholz, A.M.; Ellison, J.W.; Ravnan, J.B.; Torchia, B.S.; Ballif, B.C.; Fisher, A.J. Detection rates of clinically significant genomic alterations by microarray analysis for specific anomalies detected by ultrasound. Prenat. Diagn. 2012, 32, 986–995. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, M.; Chen, L.; Huang, H.; Xu, L. Prenatal diagnosis of BACs-on-Beads assay in 1520 cases from Fujian Province, China. Mol. Genet. Genom. Med. 2020, 8, e1446. [Google Scholar] [CrossRef]
- Breman, A.; Pursley, A.N.; Hixson, P.; Bi, W.; Ward, P.; Bacino, C.A.; Shaw, C.; Lupski, J.R.; Beaudet, A.; Patel, A.; et al. Prenatal chromosomal microarray analysis in a diagnostic laboratory; experience with >1000 cases and review of the literature. Prenat. Diagn. 2012, 32, 351–361. [Google Scholar] [CrossRef]
- Chai, H.; DiAdamo, A.; Grommisch, B.; Xu, F.; Zhou, Q.; Wen, J.; Mahoney, M.; Bale, A.; McGrath, J.; Spencer-Manzon, M.; et al. A Retrospective Analysis of 10-Year Data Assessed the Diagnostic Accuracy and Efficacy of Cytogenomic Abnormalities in Current Prenatal and Pediatric Settings. Front. Genet. 2019, 10, 1162. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Dabell, M.P.; Fisher, A.J.; Coppinger, J.; Bandholz, A.M.; Ellison, J.W.; Ravnan, J.B.; Torchia, B.S.; Ballif, B.C.; Rosenfeld, J.A. Experience with microarray-based comparative genomic hybridization for prenatal diagnosis in over 5000 pregnancies. Prenat. Diagn. 2012, 32, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Lovrecic, L.; Remec, Z.I.; Volk, M.; Rudolf, G.; Writzl, K.; Peterlin, B. Clinical utility of array comparative genomic hybridisation in prenatal setting. BMC Med. Genet. 2016, 17, 81. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Wit, M.C.; Srebniak, M.I.; Govaerts, L.C.; Van Opstal, D.; Galjaard, R.J.; Go, A.T. Additional value of prenatal genomic array testing in fetuses with isolated structural ultrasound abnormalities and a normal karyotype: A systematic review of the literature. Ultrasound Obstet. Gynecol. 2014, 43, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Sagi-Dain, L.; Maya, I.; Reches, A.; Frumkin, A.; Grinshpun-Cohen, J.; Segel, R.; Manor, E.; Khayat, M.; Tenne, T.; Banne, E.; et al. Chromosomal Microarray Analysis Results From Pregnancies With Various Ultrasonographic Anomalies. Obstet. Gynecol. 2018, 132, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Hureaux, M.; Guterman, S.; Hervé, B.; Till, M.; Jaillard, S.; Redon, S.; Valduga, M.; Coutton, C.; Missirian, C.; Prieur, F.; et al. Chromosomal microarray analysis in fetuses with an isolated congenital heart defect: A retrospective, nationwide, multicenter study in France. Prenat. Diagn. 2019, 39, 464–470. [Google Scholar] [CrossRef]
- Li, S.; Han, X.; Wang, Y.; Chen, S.; Niu, J.; Qian, Z.; Li, P.; Jin, L.; Xu, C. Chromosomal microarray analysis in fetuses with congenital anomalies of the kidney and urinary tract: A prospective cohort study and meta-analysis. Prenat. Diagn. 2019, 39, 165–174. [Google Scholar] [CrossRef]
- Hu, T.; Zhang, Z.; Wang, J.; Li, Q.; Zhu, H.; Lai, Y.; Wang, H.; Liu, S. Prenatal diagnosis of chromosomal aberrations by chromosomal microarray analysis in fetuses with ultrasound anomalies in the urinary system. Prenat. Diagn. 2019, 39, 1096–1106. [Google Scholar] [CrossRef]
- Bernier, R.; Steinman, K.J.; Reilly, B.; Wallace, A.S.; Sherr, E.H.; Pojman, N.; Mefford, H.C.; Gerdts, J.; Earl, R.; Hanson, E.; et al. Simons VIP consortium. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet. Med. 2016, 18, 341–349. [Google Scholar] [CrossRef]
- Upadhyai, P.; Amiri, E.F.; Guleria, V.S.; Bielas, S.L.; Girisha, K.M.; Shukla, A. Recurrent 1q21.1 deletion syndrome: Report on variable expression, nonpenetrance and review of literature. Clin. Dysmorphol. 2020, 29, 127–131. [Google Scholar] [CrossRef]
- Egloff, M.; Hervé, B.; Quibel, T.; Jaillard, S.; Le Bouar, G.; Uguen, K.; Saliou, A.H.; Valduga, M.; Perdriolle, E.; Coutton, C.; et al. Diagnostic yield of chromosomal microarray analysis in fetuses with isolated increased nuchal translucency: A French multicenter study. Ultrasound Obstet. Gynecol. 2018, 52, 715–721. [Google Scholar] [CrossRef]
- Chen, C.P.; Chang, S.Y.; Lin, C.J.; Chern, S.R.; Wu, P.S.; Chen, S.W.; Lai, S.T.; Chuang, T.Y.; Chen, W.L.; Yang, C.W.; et al. Prenatal diagnosis of a familial 5p14.3-p14.1 deletion encompassing CDH18, CDH12, PMCHL1, PRDM9 and CDH10 in a fetus with congenital heart disease on prenatal ultrasound. Taiwan. J. Obstet. Gynecol. 2018, 57, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Faivre, L.; Viot, G.; Prieur, M.; Turleau, C.; Gosset, P.; Romana, S.; Munnich, A.; Vekemans, M.; Cormier-Daire, V. Apparent Sotos syndrome (cerebral gigantism) in a child with trisomy 20p11.2-p12.1 mosaicism. Am. J. Med. Genet. 2000, 91, 273–276. [Google Scholar] [CrossRef]
- Li, R.; Fu, F.; Zhang, Y.L.; Li, D.Z.; Liao, C. Prenatal diagnosis of 17q12 duplication and deletion syndrome in two fetuses with congenital anomalies. Taiwan. J. Obstet. Gynecol. 2014, 53, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Nagamani, S.C.; Erez, A.; Shen, J. Clinical spectrum associated with recurrent genomic rearrangements in chromosome 17q12. Eur. J. Hum. Genet. 2010, 18, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, P.; Sen, P.; Bhatt, S.S.; Storer, M.; Xia, Z.; Bejjani, B.A.; Ou, Z.; Wiszniewska, J.; Driscoll, D.J.; Maisenbacher, M.K.; et al. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am. J. Hum. Genet. 2009, 84, 780–791, Erratum in Am. J. Hum. Genet. 2009, 85, 537. [Google Scholar] [CrossRef]
- Yu, S.; Shao, L.; Kilbride, H.; Zwick, D.L. Haploinsufficiencies of FOXF1 and FOXC2 genes associated with lethal alveolar capillary dysplasia and congenital heart disease. Am. J. Med. Genet. A 2010, 152A, 1257–1262. [Google Scholar] [CrossRef]
- Zufferey, F.; Martinet, D.; Osterheld, M.C.; Niel-Bütschi, F.; Giannoni, E.; Schmutz, N.B.; Xia, Z.; Beckmann, J.S.; Shaw-Smith, C.; Stankiewicz, P.; et al. 16q24.1 microdeletion in a premature newborn: Usefulness of array-based comparative genomic hybridization in persistent pulmonary hypertension of the newborn. Pediatr. Crit. Care Med. 2011, 12, e427–e432. [Google Scholar] [CrossRef]
- Dimitrov, B.I.; de Ravel, T.; Van Driessche, J.; de Die-Smulders, C.; Toutain, A.; Vermeesch, J.R.; Fryns, J.P.; Devriendt, K.; Debeer, P. Distal limb deficiencies, micrognathia syndrome, and syndromic forms of split hand foot malformation (SHFM) are caused by chromosome 10q genomic rearrangements. J. Med. Genet. 2010, 47, 103–111. [Google Scholar] [CrossRef]
- Bauters, M.; Frints, S.G.; Van Esch, H.; Spruijt, L.; Baldewijns, M.M.; de Die-Smulders, C.E.; Fryns, J.P.; Marynen, P.; Froyen, G. Evidence for increased SOX3 dosage as a risk factor for X-linked hypopituitarism and neural tube defects. Am. J. Med. Genet. A 2014, 164A, 1947–1952. [Google Scholar] [CrossRef]
- Hureaux, M.; Ben Miled, S.; Chatron, N.; Coussement, A.; Bessières, B.; Egloff, M.; Mechler, C.; Stirnemann, J.; Tsatsaris, V.; Barcia, G.; et al. SOX3 duplication: A genetic cause to investigate in fetuses with neural tube defects. Prenat. Diagn. 2019, 39, 1026–1034. [Google Scholar] [CrossRef]
- Wang, J.C.; Radcliff, J.; Coe, S.J.; Mahon, L.W. Effects of platforms, size filter cutoffs, and targeted regions of cytogenomic microarray on detection of copy number variants and uniparental disomy in prenatal diagnosis: Results from 5026 pregnancies. Prenat. Diagn. 2019, 39, 137–156. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Year | 2014 | 2015 | 2016 | 2017 | 2018 | 2019 | 2020 | 2021 (Until to the Day 30 September 2021) | Total Results |
|---|---|---|---|---|---|---|---|---|---|
| All Results | 71 | 195 | 398 | 733 | 1191 | 1710 | 1812 | 1290 | 7400 |
| Normal | 51 | 142 | 276 | 522 | 844 | 1163 | 1290 | 976 | 5264 (71.6%) |
| Abnormal | 17 | 51 | 110 | 190 | 321 | 539 | 495 | 287 | 2010 (26.7%) |
| Non-informative | 3 | 2 | 12 | 21 | 16 | 12 | 33 | 27 | 126 (1.7%) |
| Indications | Number of Patients | Normal Results | Abnormal Results | Aneuploidy and Triploidy | CNVs | P | LP | VOUS | LB | Non- Informative Results | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Achondroplasia | 3 | 3 | 0 | 0.0% | 0 | 0 | - | - | - | - | 0 |
| Gastroschisis | 7 | 6 | 0 | 0.0% | 0 | 0 | - | - | - | - | 1 |
| Parental Anxiety | 13 | 13 | 0 | 0.0% | 0 | 0 | - | - | - | - | 0 |
| Skeletal defects | 65 | 54 | 10 | 15.4% | 6 | 4 | 2 | 2 | - | - | 1 |
| Omphalocele | 69 | 52 | 13 | 18.8% | 8 | 5 | 2 | 1 | 2 | - | 4 |
| Fetal hypotrophy | 83 | 60 | 19 | 22.9% | 12 | 7 | 7 | - | - | - | 4 |
| Hydrops fetalis | 123 | 61 | 59 | 48.0% | 44 | 15 | 15 | - | - | - | 3 |
| Cranio-facial defects | 133 | 111 | 22 | 16.5% | 18 | 4 | 4 | - | - | - | 0 |
| Urinary system defects | 134 | 124 | 6 | 4.5% | 3 | 3 | 1 | 2 | - | - | 4 |
| Central nervous system | 154 | 121 | 31 | 20.1% | 16 | 15 | 8 | 3 | 3 | 1 | 2 |
| Genetic disorders in family history | 207 | 137 | 60 | 29.0% | 31 | 29 | 21 | 4 | 1 | 3 | 10 |
| Advanced Maternal Age | 336 | 256 | 70 | 20.8% | 52 | 18 | 8 | 10 | - | - | 10 |
| NT | 573 | 345 | 212 | 36.9% | 199 | 13 | - | - | 4 | 4 | 16 |
| Abnormal results of biochemical test PAPP-A | 713 | 601 | 104 | 14.6% | 82 | 22 | 14 | 8 | 6 | - | 8 |
| Congenital heart defects | 1188 | 894 | 285 | 24.0% | 162 | 123 | 94 | 8 | 21 | - | 9 |
| Verification of the others tests | 1354 | 918 | 418 | 30.9% | 309 | 109 | 104 | 2 | - | 3 | 18 |
| Multiple birth defects | 2245 | 1508 | 701 | 31.2% | 489 | 212 | 166 | 19 | 20 | 6 | 36 |
| Total results | 7400 | 5264 | 2010 | 27.2% | 1431 | 579 | 446 | 59 | 57 | 17 | 126 |
| Age | Number of Patients | Normal Results | Abnormal Results | Aneuploidy and Triploidy | Pathogenic Structural Abnormality | |||
|---|---|---|---|---|---|---|---|---|
| 35–39 | 191 | 153 | 38 | 19.8% | 25 | 13.1% | 13 | 6.8% |
| 40–44 | 119 | 93 | 26 | 21.8% | 21 | 17.6% | 5 | 4.2% |
| 45–49 | 26 | 20 | 6 | 23.1% | 6 | 23.1% | 0 | 0.0% |
| Total results | 336 | 266 | 70 | 20.8% | 52 | 15.5% | 18 | 5.4% |
| Number of Patients | Normal Results | Abnormal Results | Trisomy of a Specific Chromosome | Monosomy X | Structural Aberrations | ||||
|---|---|---|---|---|---|---|---|---|---|
| High risk of T21 | 615 | 514 | 101 | 78 | 12.7% | 6 | 1.0% | 17 | 2.8% |
| High risk of T18 | 36 | 22 | 14 | 13 | 36.1% | - | - | 1 | 2.8% |
| High risk of T13 | 20 | 15 | 5 | 5 | 25.0% | - | - | 0 | 0.0% |
| High risk of T21, T18, T13 | 42 | 30 | 12 | 2xT13, 3xT18, 3xT21 | 19.0% | - | - | 4 | 9.5% |
| Total results | 713 | 581 | 132 | 104 | 14.6% | 6 | 0.8% | 22 | 3.1% |
| Patient Number | High Risk of Trisomy | aCGH Results |
|---|---|---|
| 1362 | T21 | 21q21.1q22.11(19352022_33657166)x3, 21q22.11q22.13(35734654_38189781)x3, 21q22.2(41187502_41429023)x3, 21q22.3(42850375_46139875)x3, 21q22.3(46991188_47561714)x3 |
| 1985 | T21 | Xp22.33p11.23(76118_46742615)x1 |
| 2145 | T21 | 22q11.21(18847961_21457610)x3 |
| 2219 | T21 | 9p22.3(14747397_15041021)x3 |
| 2652 | T21, T18, T13 | 2q22.1q35(142089727_219021345)x3 |
| 3356 | T21 | 1p32.3(50817235_52280457)x3 pat |
| 3463 | T21 | 4q32.1q32.2(160399605_163122660)x3 pat |
| 3476 | T21 | 22q11.21(18894820_21457610)x3 |
| 3769 | T21 | Xp11.22(53463247_53790726)x3 |
| 4099 | T21 | 1q21.1q21.2(146155929_147824212)x1 |
| 5342 | T21 | 22q11.21(21081284_21457610)x1 dn |
| 5672 | T21, T18, T13 | 5p15.33p15.32(22149_4768822)x1, 5p15.2(10212960_12513658)x1, 11p15.5p14.1(113082_27880946)x3 |
| 6139 | T21 | 1p13.2p13.1(115761998_116816569)x3 |
| 6219 | T21 | 16p12.2(21926361_22407951)x1 mat |
| 7121 | T21 | 20p12.3p11.1(5593060_25678293)x3 |
| 7408 | T21 | 5p14.3p14.1(19364195_25086222)x1 |
| Case Number | Aberration | Indications for the Study | Size | Sex | Inheritance | Interpretation |
|---|---|---|---|---|---|---|
| 1956 | 17q12(34569737_36290311)x3 | cyst in the fetus | 1.72 Mb | F | unknown | The aberration covers the region of the known RCAD syndrome (OMIM: 137920), described in patients with MODY diabetes and renal cystic disease. Duplications in the 17q12 region can manifest themselves in a wide spectrum of clinical symptoms. |
| 2652 | 2q22.1q35(142089727_219021345)x3 | screening test showing high risk of chromosomal aberration T 18 1:5, T21 1:5 | 77 Mb | F | unknown | The aberration spans multiple genes and is responsible for the abnormalities found in the fetus in ultrasound examination. |
| 3426 | Xp21.1(32006239_32383121)x0 | NT = 3mm | 377 kb | M | unknown | The deletion includes exons 36–44 of the DMD gene (OMIM 300377). Mutations, deletions of this gene have been reported in patients with Duchenne Muscular Dystrophy (OMIM: 310200) and Becker Muscular Dystrophy (OMIM: 300376). |
| 5027 | 15q13.2q13.3(30389992_32702923)x1 | mitral valve atresia, left ventricular hypoplasia, aortic hypoplasia | 2.31 Mb | M | unknown | Deletions of this region have been described in patients mainly with psychomotor retardation, epilepsy, behavioral disorders, less often with discrete dysmorphic features and congenital heart defects (ORPHA: 199318) |
| 5037 | 16q23.1(75598893_78008020)x1, 17p12(14111754_15442178)x1 | hypotrophy, cleft lip | 2.41 Mb; 1.33 Mb | F | unknown | The deletion in the 16q23.1 region includes 10 genes encoding proteins: GABARAPL2 (OMIM 607452), ADAT1 (OMIM 604230), KARS (OMIM 601421), TERF2IP (OMIM 605061), CNTNAP4 (OMIM 610518), MON1B (OMIM 608954), SYCE1L2, ADAMTS18 (OMIM 607512), NUDT7 (OMIM 609231), VAT1L2. The deletion 17p12 region includes 7 genes encoding proteins: COX10 (OMIM 602125), CDRT152, HS3ST3B1 (OMIM 604058), dose-sensitive PMP22 gene (OMIM 601097), TEKT3 (OMIM 612683), CDRT42, TVP23C2 and the critical region of hereditary neuropathy syndrome with hypersensitivity to pressure HNPP (OMIM 162500). |
| 5356 | 16p12.2(21926361_22645787)x1 | screening test showing high risk of chromosomal aberration (T21 1: 18) | 720 kb | M | unknown | Additionally, the study revealed a deletion of the short arm of chromosome 16 in the 16p12.2 region of ~720 kb. The aberration covers the region of the known 16p12 deletion syndrome (OMIM 136570), described in patients including with: intellectual disability/psychomotor retardation, congenital heart defects and craniofacial dysmorphic features. |
| 5500 | 5q35.2q35.3(175116131_180696832)x1 | NT = 7.0 mm, heart disease | 5.58 Mb | F | unknown | The deletion includes 87 protein-coding genes, including the NSD1 gene (OMIM 606681) and is located in the region of the 5q35 deletion syndrome (ORPHA: 1627), characterized by: lymphoedema with enlargement of the nuchal translucency in the prenatal period, as well as early childhood hypotension, low height, facial dysmorphic features and heart defects. The 5q35 deletion region includes the region of the Sotos 1 complex (SOTOS1, OMIM 117550), described in patients with: facial dysmorphic features, high birth weight and excess growth early in life, macrocephaly, intellectual disability, as well as heart defects such as atrial septal defect (ASD), ventricular septal defect (VSD). |
| 5653 | 17q12(34569737_36326421)x3, Xq28(153884022_154491017)x3 | defect of the central nervous system | 1.76 Mb; 607kb | F | de novo; paternal | Duplication in the 17q12 region includes 21 protein-coding genes, including genes: CCL3L1 (OMIM 601395), ZNHIT3 (OMIM 604500) 1, PIGW (OMIM 610275) and dose-sensitive genes: LHX1 (OMIM 601999), AATF (OMIM 608463), ACACA (OMIM 200350), TADA2A (OMIM 602276), HNF1B (OMIM 189907) and the duplication syndrome region 17q12 (OMIM 614526). Duplication in the Xq28 region includes 13 protein-coding genes: GAB3 (OMIM 300482), dose-sensitive DKC1 (OMIM 300126), MPP1 (OMIM 305360), SMIM96, F8 (OMIM 300841), H2AFB1 (OMIM 301037), F8A1 (OMIM 305423), FUNDC2 (OMIM 301042), CMC46, MTCP1 (OMIM 300116), BRCC3 (OMIM 300617), VBP1 (OMIM 300133) and exon 2 of the RAB39B gene (OMIM 300774) and partially the region Xq28 distal microduplication syndrome (ORPHA: 293939) |
| 5843 | Xp22.33(76118_1625396)x0 or Yp11.32(27254_1570153)x0, Yq11.221q12(17751498_59329063)x0 | screening test showing high risk of chromosomal aberration (T21 = 1:19, T13 = 1:44, T18 = 1:60) | 1,55 Mb; 41,58 Mb | M | unknown | The deletion of the Xp22.33 or Yp11.32 region includes 10 protein-coding genes including the CSF2RA (OMIM 306250, OMIM: 425000) and SHOX (OMIM: 312865, OMIM: 400020) genes. SHOX deletions are pathogenic changes and have been reported in idiopathic short stature patients (OMIM: 300582) (ORPHA: 314795). The identified aberrations indicate the presence of an abnormal Y chromosome. |
| 6219 | 16p12.2(21926361_22407951)x1 | screening test showing high risk of chromosomal aberration (T21 1: 54) | 482 kb | F | maternal | The aberration covers the region of the known 16p12 deletion syndrome (OMIM 136570), including 8 genes encoding proteins: UQCRC2 (OMIM 191329), PDZD92, C16orf522, VWA3A2, SDR42E22, EEF2K (OMIM 606968), POLR3E (OMIM 617815) and CDR3E (OMIM 117340). Deletions in the 16p12 region were described in patients with intellectual disability/psychomotor retardation, congenital heart defects and craniofacial dysmorphic features. |
| 6420 | 15q22.31q26.3(63965478_102383479)x3 | brain defect | 38.42 Mb | F | unknown | The duplication involves 283 genes encoding the protein, including the 15q overgrowth syndrome (ORPHA: 314585). Partial trisomy 15q has been reported in patients including with facial dysmorphic features, excessive pre- and postnatal growth, kidney defects (e.g., agenesis, hydronephrosis), school difficulties/intellectual disability and behavioral disorders. Additionally, craniosynostosis and macrocephaly are described. |
| 6527 | 12q24.21(114791285_114846644)x1 | AVSD, reduced aortic dimensions | 55 kb | M | unknown | The deletion includes the dose sensitive TBX5 gene (OMIM 601620). Deletions and mutations of this gene have been reported in patients with HOS type 1 syndrome (Holt–Oram syndrome; OMIM 142900) with congenital heart defects and malformations of the upper limbs. |
| 6678 | (18)x3,Xq28(154199319_154560374)x3 | age 42, cleft palate | trisomy 18; 361 kb | F | unknown | Edwards syndrome. Additionally, the study found duplication in the Xq28 region. The duplication includes 8 genes encoding proteins: exons 1–6 of the F8 gene (OMIM 300841), FUNDC2 genes (OMIM 301042), CMC42, MTCP1 (OMIM 300116), BRCC3 (OMIM 300617), VBP1 (OMIM 300133), RAB39B (OMIM 300774) and exons 2–6 of the CLIC2 gene (OMIM 300138). The aberration covers the region of distal microduplication syndrome Xq28 (OMIM 300815) (ORPHA: 293939). |
| 7030 | 17p13.3(736836_1227471)x3 | cerebral hernia, VSD, clubfoot | 491 kb | F | maternal | The duplication includes exon 1 of the NXN gene (OMIM 612895) and the genes TIMM22 (OMIM 607251), ABR (OMIM 600365), BHLHA9 (OMIM 615416), TRARG1 (OMIM 612211). Duplications involving the BHLHA9 gene have been reported in patients with long bone aplasia-associated cleft hand/foot type 3 (SHFLD3) (OMIM 612576) and in patients with a cleft femur-mesomial ectrodactyly (OMIM 228250) (ORPHA: 1986). Less frequently, these patients may develop heart defects, cleft lip and palate, and esophageal atresia2. Duplications of this region are characterized by variable expression and incomplete penetrance (less than 50%). |
| 7271 | 16q24.1(86053209_86705830)x1 | age 35, screening test showing high risk of chromosomal aberration (T21 1:29), multiple defects | 653 kb | M | de novo | The deletion includes 4 protein-encoding genes: dose-sensitive FOXF1 gene (OMIM 601089), MTHFSD gene (OMIM 616820), dose-sensitive FOXC2 gene (OMIM 602402), FOXL1 gene (OMIM 603252) and is located in the band region 16q24.1 microdeletion (ORPHA: 352629). Deletions in the 16q24.1 region involving the FOX gene cluster are described in patients, among others with vascular dysplasia of the alveoli with displacement of pulmonary vessels, heart defects, gastrointestinal defects and urinary tract defects, including hydronephrosis. |
| 7296 | 16p13.3(3788560_3808214)x1 | hypotrophy, NT = 3.5 mm | 20 kb | F | unknown | The deletion includes exons 18–26 of the dose-sensitive CREBBP gene (OMIM 600140). Deletions and mutations of this gene are described in patients with Rubinstein–Taybi syndrome type 1 (OMIM 180849), who have, among others short stature, postnatal growth retardation, heart defects, skeletal system defects, microcephaly, intellectual disability and dysmorphic features face. |
| Case Number | Aberration | Indications for the Study | Size | Sex | Inheritance | Interpretation |
|---|---|---|---|---|---|---|
| 1431 | 11p11.2(43713787_45797075)x3 | hypertelorism, asymmetry of the width of the lateral ventricles of the brain, hypoplastic bone of the nose | 2.08 Mb | F | maternal | The duplication covers a region of the known 11p11.2 microdeletion (Syndrom Potocki-Shaffer, OMIM 601224). |
| 2558 | 7p14.1(42058801_42738664)x3 | hydrothorax, IUGR | 69 kb | F | maternal | The aberration includes exons 1–10 of the dose-sensitive GLI3 gene (OMIM: 165240), the mutations, deletions and duplications of which have been reported in patients with Greig cephalopolysindactyly syndrome (ang. Greig cephalopolysyndactyly syndrome, GCPS) (OMIM: 175700). |
| 2643 | Xq27.1(139283433_139743154)x3, 10q24.31q24.32(102880054_ 103538415)x3 | age 37, cerebral hydrocephalus, the width of the lateral ventricle 13 mm, concave outline of the frontal bones (symptom of lemon and banana), hernia in the sacro-lumbar section | 658 kb; 565 kb | F | maternal | Duplication in the 10q24.31q24.3 region includes the following genes: BTRC (OMIM 603482), DPCD (OMIM 616467), FBXW4 (OMIM 608071), as well as exons 1–2 of the TLX1NB gene (OMIM 612734) and dose-sensitive genes: TLX1 (OMIM 186770), LBX1 (OMIM 604255), POLL (OMIM 606343), and FGF8 (OMIM 600483). Duplications of this region have been reported in patients with limb defects (SHFM3; OMIM 246560) and may be inherited from the parents. Duplication in the Xq27.1 2 region involves the dose-sensitive SOX3 gene (OMIM 313430). Duplications of this region have been reported in patients with polyhormonal hypopituitarism (OMIM 312000), sex-linked intellectual disability with isolated growth hormone deficiency (OMIM 300123), and neural tube defects. |
| 3377 | 16q24.3(89804031_89897059)x1 | NT = 4 mm, agenesis of the corpus callosum | 93 kb | M | unknown | The deletion includes exons 10–11 of the ZFP276 gene (OMIM: 608460), exon 1 of the SPIRE2 gene (OMIM: 609217) and the FANCA gene (OMIM: 607139). FANCA gene mutations and deletions have been reported in patients with Group A Fanconi Anemia (OMIM: 227650) |
| Case Number | Aberration | Size | Gene/Genes | Indications |
|---|---|---|---|---|
| 48 | 9q33.2(124229923_124370633)x3 | 140.71 kb | OR1J1 | omphalocele, cleft lip and palate |
| 67 | 7q31.1(110410697_110836614)x1 | 425.92 kb | intron 3–4 IMMP2L, LRRN3 | NT 7.5 mm, micrognathia, fetal heart defect |
| 193 | 1q42.2(230763393_231441324)x3, 2q21.1(129829959_131404737)x1 | 677 kb; 1.57 Mb | PCNXL2, ARHGEF4 | generalized fetal swelling, NT = 3.8 mm, no NB, flat face profile |
| 264 | Xp11.4(38096994_38138665)x3 | 41 kb | OTC | family history |
| 303 | 6q14.3q15(86892972_87919594)x3 | 1.03 Mb | HTR1E | NT = 4 mm, cleft lip |
| 395 | 5q35.3(177068821_178058571)x3 | 989 kb | PROP1, NHP2 | hypotrophy, VSD |
| 584 | 13q13.3(37145323_37351415)x3 | 206 kb | SERTM1 | cardiac ectopy |
| 601 | 7q35(146544277_146840480)x1 | 296 kb | ex 4–8 CNTNAP2 | cleft lip, hypotrophy, ASD |
| 602 | 7q35(146544277_146840480)x1 | 296 kb | ex 4–8 CNTNAP2 | widening of the lateral ventricles of the brain, VSD |
| 608 | 4p15.32(16064173_16813206)x3 | 750 kb | TAPT1, PROM1, LDB | AVSD |
| 609 | 4p15.32(16064173_16813206)x3 | 750 kb | TAPT1, PROM1, LDB | AVSD |
| 674 | 2p16.3(48059806_48500445)x3 | 440 kb | ex 1–10 FBXO11 | tricuspid valve regurgitation |
| 674 | 2p16.3(48059806_48500445)x3 | 440 kb | FBXO11 | tricuspid valve regurgitation |
| 730 | 8p23.1(11607828_11723203)x3 | 115 kb | GATA4, NEIL2, FDFT1, CTSB | NT |
| 765 | 11q22.1(101436248_101756583)x3 | 320 kb | ex 1 TRPC6 | atrioventricular septal defect (AVSD) |
| 845 | 2q23.1(149008939_149099960)x1 | 90 kb | ex 5 MBD5 | AVSD, duodenal obstruction |
| 851 | 18q11.1(18542080-18672140)x1 | 130 kb | ROCK1 | omphalocele, NT = 4.2 mm |
| 900 | 8p22(15570685_16812645)x1 | 1.24 Mb | MSR1, TUSC3 | cystic hygroma, gastroschisis |
| 913 | 22q11.21(19338815_19584890)x3 | 246 kb | HIRA, CCDC45, UFD1L | TOF |
| 924 | 14q32.11(91122067_91681738)x3 | 559 kb | TTC7B, RPS6KA5 | omphalocele |
| 963 | 5p15.33(95276_220479)x3 | 125 kb | PLEKHG4B, LRRC14B | TOF and no thymus |
| 1045 | 21q11.2(15824276_16137741)x3 | 313 kb | SAMSN1 | Ebstein Syndrome |
| 1093 | 13q31.3(92065636_92299097)x3 | 233 kb | ex 2 GCP5 | ARSA |
| 1165 | 9q21.32q21.33(86825588_87161409)x3 | 335 kb | SLC28A3 | CAT |
| 1198 | 1q31.1q31.2(188762960_192117536)x3 | 3.35 Mb | FAM5C | abnormal results of PAPP-A test T21 1:8 |
| 1278 | 1p36.32(2633351_3161118)x3 | 522 kb | ex 1–3 PRDM16 | abnormal heart rotation |
| 1280 | 2q14.2(121549137_121659393)x3 | 110 kb | GLI2 | AVSD |
| 1641 | 16q24.3(89584335_90252496)x3 6 | 668 kb | TUBB3 | corpus callosum agensy |
| 1658 | Xp22.2(11600766_12080374)x2 | 480 kb | MSL3, ex 1 ARHGAP6 | CHD |
| 1776 | 2q11.1q11.2(96766564_97643367)x3 | 876 kb | NCAPH, SEMA4C | Dandy-Walker syndrome |
| 1889 | 8q24.11(118391317_118716415)x3 | 325 kb | MED30 | omphalocele |
| 1969 | Xq22.3(105159857_105621192)x2 | 460 kb | SERPINA7, ex 16–29 NRK | NT 4 mm |
| 2033 | 2p16.3(50831229_50883635)x1 | 52 kb | ex 4–8 NRXN1 | hydrocephalus, pyelectasia. |
| 2089 | 2q31.1(172529289_172676299)x3 | 147 kb | ex 10–18 SLC25A12 | cleft lip, VSD |
| 2093 | 10q26.12(122509983_122668106)x3 | 158 kb | WDR11 | Ebstein Syndrome |
| 2133 | 18q21.31(54832550_55998895)x3 | 1.17 Mb | ST8SIA3, ONECUT2, FECH, NARS, ATP8B1, ex 1–11 NEDD4L | agenesis of the corpus callosum, concavity of the frontal bones |
| 2195 | Xq28(153324080_153362472)x2 | 30 kb | ex 2 MECP2 | CHD |
| 2679 | 5q22.2(112062907_112440503)x3 | 377 kb | APC, DCP2, MCC, REEP5, SRP19 | abdominal cyst |
| 2715 | 16q23.3q24.1(84115545_84899135)x3 | 784 kb | COLT1, DNAAF1, MBTPS1, USP10 | VSD, renal pyelectasia, PAPP-A test abnormal: intermediate risk of T21 1:10 |
| 2824 | 16p13.11(16041699_16311080)x3 | 269 kb | ABCC1, ABCC6 | abdominal tumor in the fetus, suspicion of Central Nervous System bleeding. |
| 2889 | 7p14.2(35241982_35280550)x1 | 39 kb | ex 5–8 TBX20 | hyperechoic gut, choroidal plexus cysts. |
| 2909 | 9p24.3(224412_381572)x1 | 157 kb | ex 2–21 DOCK8 | T21 1:56 |
| 3011 | 7q33(137363460_137560000)x3 | 196 kb | ex 1–2 DGKI | CHD |
| 3267 | 5q35.1(172105222_172352411)x1 | 247 kb | ex 1–6 ERGIC1, ex 3–5 NEURL1B, DUSP1 | hydrocephalus |
| 3356 | 1p32.3(50817235_52280457)x3 | 1.46 Mb | FAF1, CDKN2C, EPS15, OSBPL9, ex 14–33 NRD1 | IUGR, VSD |
| 3403 | 4p15.31(20421696_20673992)x1 | 252 kb | ex 5–37 SLIT2 | FGR, hypoplastic NB, shortening of the bones of the long limbs |
| 3433 | 20q13.33(58725829_60112343)x1 | 1.39 Mb | ex 1–2 CDH4, | NT 4.7 mm |
| 3488 | 9q33.1(119029804_119319068)x3 | 289 kb | ex 9–22 PAPPA, 19–22 ASTN2 | VSD |
| 5605 | 6p21.1(42824821_43112733)x3 | 288 kb | ex 10–13 GLTSCR1L, RPL7L1, PTCRA, CNPY3, GNMT, PEX6, PPP2R5D, MEA1, KLHDC3, RRP36, CUL7, MRPL2, ex 1–15 PTK7 | TOF |
| 5764 | 3p26.3(1539221_2757051)x3 | 1.22 Mb | ex 1–4 CNTN4 | abnormal results of PAPP-A test T21 1:16 |
| 6428 | 16q23.3(82563542_83763740)x1 | 1.2 Mb | ex 1–11 CDH13 | AVSD |
| 6874 | 6q22.31(123668064_124141121)x3 | 474 kb | ex 1 NKAIN2 | abnormal results of PAPP-A test T21 1:45 |
| 7055 | 1p12(120451037_120520297)x1 | 69 kb | ex 6–34 NOTCH2 | TOF |
| 7192 | 11p15.2(14696412_15028562)x1 | 332 kb | ex 2–16 PDE3B, CYP2R1, CALCA, CALCB | NT = 4.0 mm, intestine hyperechoic |
| 7209 | 15q26.3(100569119_100666644)x1 | 97 kb | ex 13–18 ADAMST17 | AVSD, cleft palate |
| 7356 | 1q43(236761288_236926498)x3 | 165 kb | ex 1–4 HEATR1, ACTN2 | age, abnormal results of PAPP-A test T21 1:15 |
| 7364 | 16q23.1q23.2(78658360_79489094)x3 | 830 kb | ex 9 WWOX | abnormal results of PAPP-A test T21 1:43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalczyk, K.; Bartnik-Głaska, M.; Smyk, M.; Plaskota, I.; Bernaciak, J.; Kędzior, M.; Wiśniowiecka-Kowalnik, B.; Deperas, M.; Domaradzka, J.; Łuszczek, A.; et al. Comparative Genomic Hybridization to Microarrays in Fetuses with High-Risk Prenatal Indications: Polish Experience with 7400 Pregnancies. Genes 2022, 13, 690. https://doi.org/10.3390/genes13040690
Kowalczyk K, Bartnik-Głaska M, Smyk M, Plaskota I, Bernaciak J, Kędzior M, Wiśniowiecka-Kowalnik B, Deperas M, Domaradzka J, Łuszczek A, et al. Comparative Genomic Hybridization to Microarrays in Fetuses with High-Risk Prenatal Indications: Polish Experience with 7400 Pregnancies. Genes. 2022; 13(4):690. https://doi.org/10.3390/genes13040690
Chicago/Turabian StyleKowalczyk, Katarzyna, Magdalena Bartnik-Głaska, Marta Smyk, Izabela Plaskota, Joanna Bernaciak, Marta Kędzior, Barbara Wiśniowiecka-Kowalnik, Marta Deperas, Justyna Domaradzka, Alicja Łuszczek, and et al. 2022. "Comparative Genomic Hybridization to Microarrays in Fetuses with High-Risk Prenatal Indications: Polish Experience with 7400 Pregnancies" Genes 13, no. 4: 690. https://doi.org/10.3390/genes13040690
APA StyleKowalczyk, K., Bartnik-Głaska, M., Smyk, M., Plaskota, I., Bernaciak, J., Kędzior, M., Wiśniowiecka-Kowalnik, B., Deperas, M., Domaradzka, J., Łuszczek, A., Dutkiewicz, D., Kozar, A., Grad, D., Niemiec, M., Ziemkiewicz, K., Magdziak, R., Braun-Walicka, N., Barczyk, A., Geremek, M., ... Nowakowska, B. A. (2022). Comparative Genomic Hybridization to Microarrays in Fetuses with High-Risk Prenatal Indications: Polish Experience with 7400 Pregnancies. Genes, 13(4), 690. https://doi.org/10.3390/genes13040690