
Development and Optimization of a Silica Column-Based Extraction Protocol for Ancient DNA

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Information
2.2. Sampling
2.3. Extraction Experiment
2.3.1. Predigestion
2.3.2. Bleach Wash and Predigestion
2.3.3. Silica Columns
2.3.4. Centrifugal Concentrator Filters
2.3.5. USER
2.4. Library Preparation and Sequencing
2.5. Data Processing and Analysis
3. Results
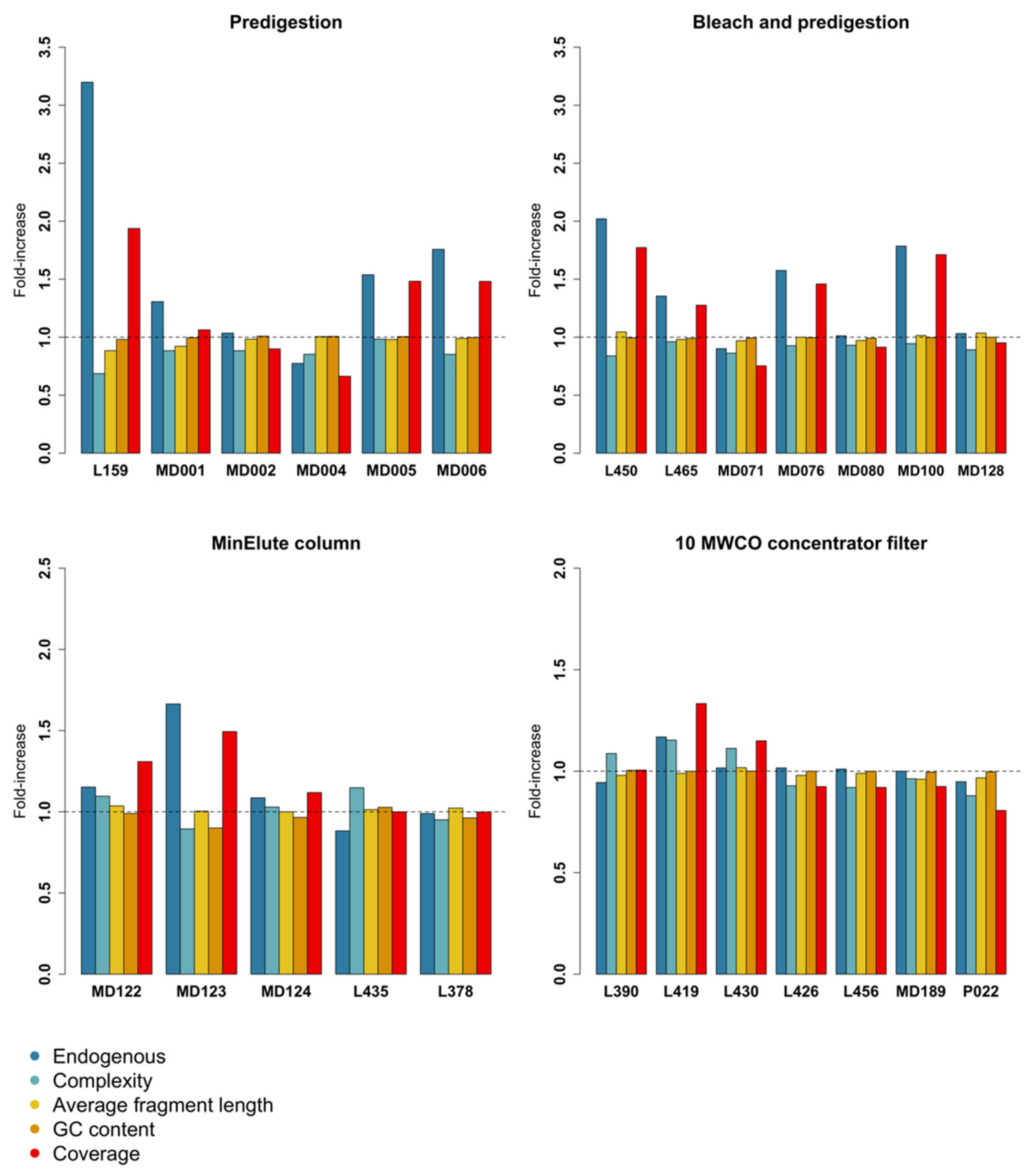
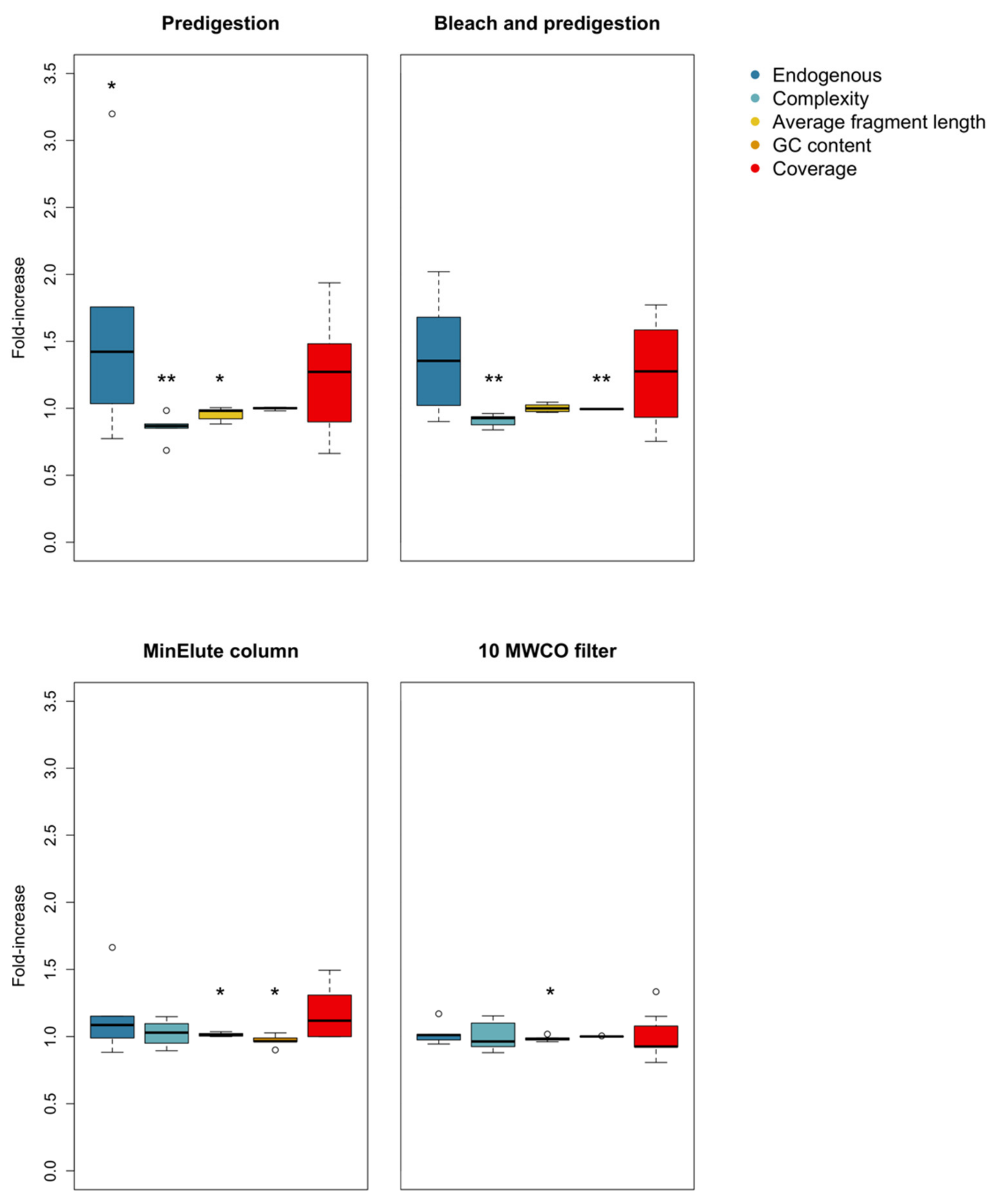
3.1. Predigestion
3.2. Bleach and Predigestion
3.3. Silica Columns
3.4. Centrifugal Concentrator Filters
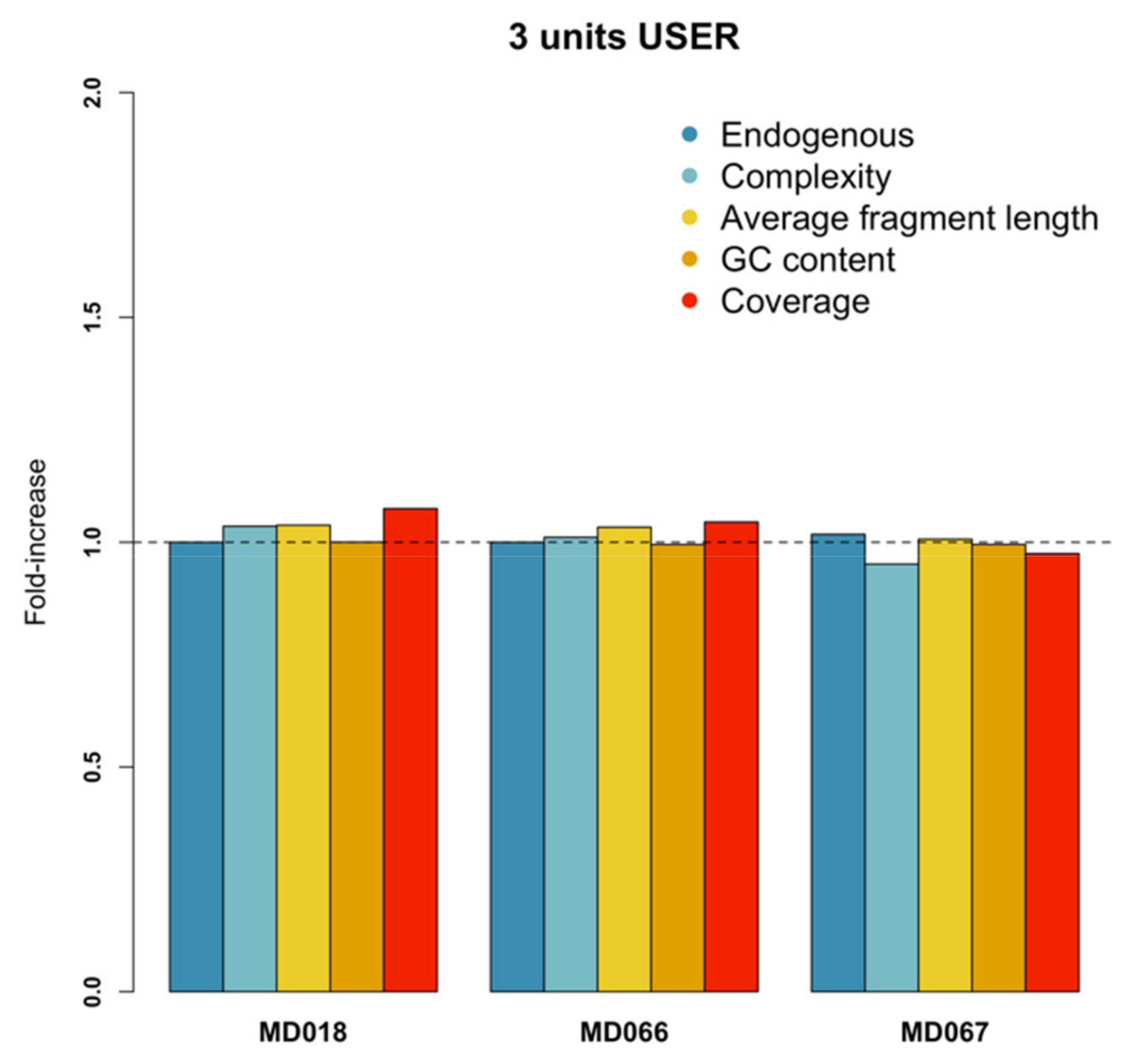
3.5. USER
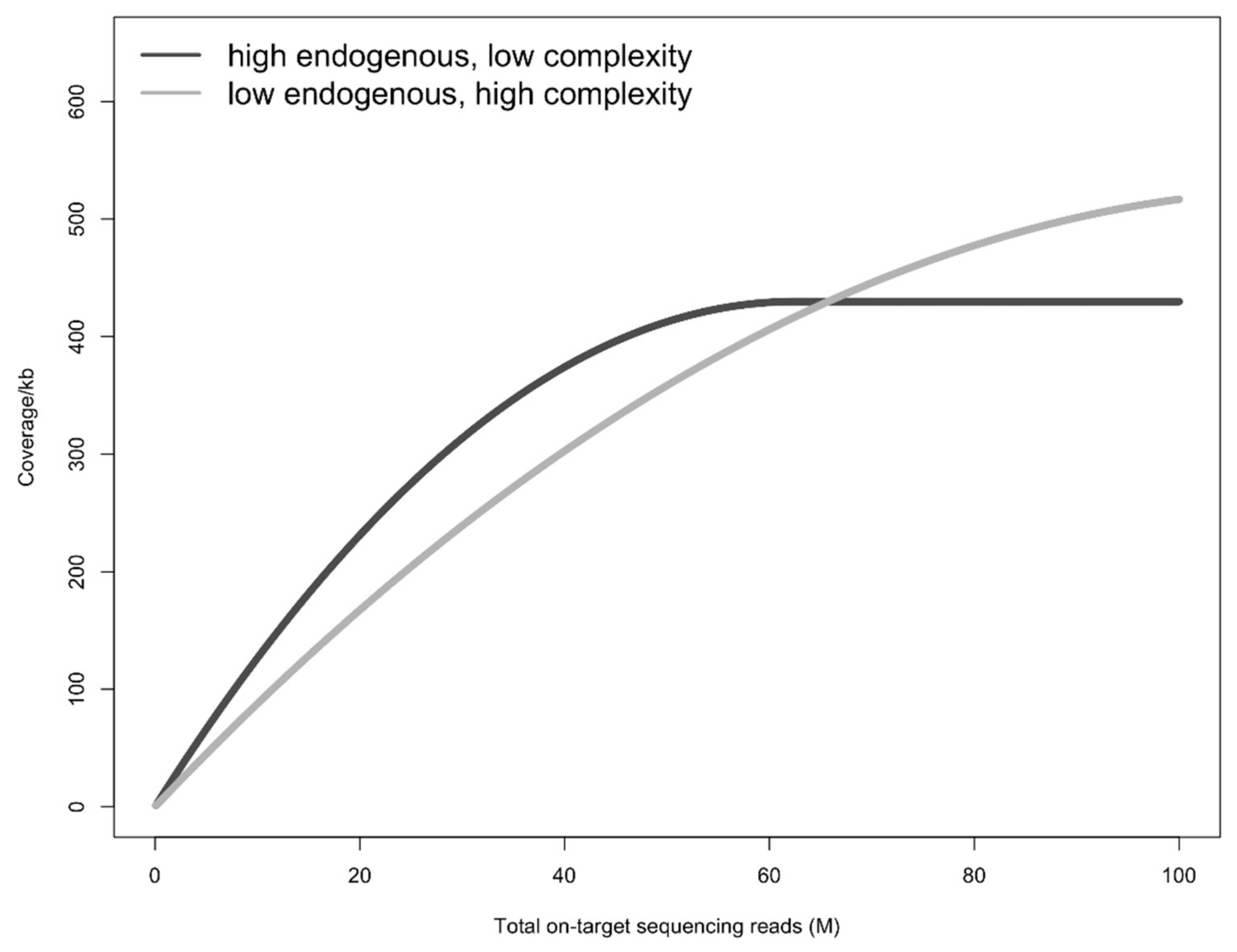
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shapiro, B.; Hofreiter, M. A Paleogenomic Perspective on Evolution and Gene Function: New Insights from Ancient DNA. Science 2014, 343, 1236573. [Google Scholar] [CrossRef] [PubMed]
- Burrell, A.S.; Disotell, T.R.; Bergey, C.M. The Use of Museum Specimens with High-Throughput DNA Sequencers. J. Hum. Evol. 2015, 79, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, M.; Librado, P.; Der Sarkissian, C.; Schubert, M.; Alfarhan, A.H.; Alquraishi, S.A.; Al-Rasheid, K.A.S.; Gamba, C.; Willerslev, E.; Orlando, L. Evolutionary Patterns and Processes: Lessons from Ancient DNA. Syst. Biol. 2017, 66, e1–e29. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M.H.-Y.; et al. A Draft Sequence of the Neandertal Genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Palkopoulou, E.; Mallick, S.; Skoglund, P.; Enk, J.; Rohland, N.; Li, H.; Omrak, A.; Vartanyan, S.; Poinar, H.; Götherström, A.; et al. Complete Genomes Reveal Signatures of Demographic and Genetic Declines in the Woolly Mammoth. Curr. Biol. 2015, 25, 1395–1400. [Google Scholar] [CrossRef]
- Allentoft, M.E.; Sikora, M.; Sjögren, K.-G.; Rasmussen, S.; Rasmussen, M.; Stenderup, J.; Damgaard, P.B.; Schroeder, H.; Ahlström, T.; Vinner, L. Population Genomics of Bronze Age Eurasia. Nature 2015, 522, 167. [Google Scholar] [CrossRef]
- Haak, W.; Lazaridis, I.; Patterson, N.; Rohland, N.; Llamas, B.; Brandt, G.; Nordenfelt, S.; Harney, E.; Stewardson, K.; Fu, Q.; et al. Massive Migration from the Steppe Was a Source for Indo- European Languages in Europe. Nature 2015, 522, 207–211. [Google Scholar] [CrossRef]
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; García, N.; Pääbo, S.; Arsuaga, J.L.; et al. Complete Mitochondrial Genome Sequence of a Middle Pleistocene Cave Bear Reconstructed from Ultrashort DNA Fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758–15763. [Google Scholar] [CrossRef]
- Orlando, L.; Ginolhac, A.; Zhang, G.; Froese, D.; Albrechtsen, A.; Stiller, M.; Schubert, M.; Cappellini, E.; Petersen, B.; Moltke, I. Recalibrating Equus Evolution Using the Genome Sequence of an Early Middle Pleistocene Horse. Nature 2013, 499, 74–78. [Google Scholar] [CrossRef]
- Van der Valk, T.; Pečnerová, P.; Díez-del-Molino, D.; Bergström, A.; Oppenheimer, J.; Hartmann, S.; Xenikoudakis, G.; Thomas, J.A.; Dehasque, M.; Sağlıcan, E.; et al. Million-Year-Old DNA Sheds Light on the Genomic History of Mammoths. Nature 2021, 591, 265–269. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and Decay of the Primary Structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Allentoft, M.E.; Collins, M.; Harker, D.; Haile, J.; Oskam, C.L.; Hale, M.L.; Campos, P.F.; Samaniego, J.A.; Gilbert, T.P.M.; Willerslev, E.; et al. The Half-Life of DNA in Bone: Measuring Decay Kinetics in 158 Dated Fossils. Proc. R. Soc. B Biol. Sci. 2012, 279, 4724–4733. [Google Scholar] [CrossRef]
- Kistler, L.; Ware, R.; Smith, O.; Collins, M.; Allaby, R.G. A New Model for Ancient DNA Decay Based on Paleogenomic Meta-Analysis. Nucleic Acids Res. 2017, 45, 6310–6320. [Google Scholar] [CrossRef]
- Der Sarkissian, C.; Ermini, L.; Jónsson, H.; Alekseev, A.N.; Crubezy, E.; Shapiro, B.; Orlando, L. Shotgun Microbial Profiling of Fossil Remains. Mol. Ecol. 2014, 23, 1780–1798. [Google Scholar] [CrossRef]
- Pääbo, S. Ancient DNA: Extraction, Characterization, Molecular Cloning, and Enzymatic Amplification. Proc. Natl. Acad. Sci. USA 1989, 86, 1939–1943. [Google Scholar] [CrossRef]
- Stiller, M.; Green, R.E.; Ronan, M.; Simons, J.F.; Du, L.; He, W.; Egholm, M.; Rothberg, J.M.; Keates, S.G.; Ovodov, N.D. Patterns of Nucleotide Misincorporations during Enzymatic Amplification and Direct Large-Scale Sequencing of Ancient DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 13578–13584. [Google Scholar] [CrossRef]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.F.; Green, R.E.; Kelso, J.; Prüfer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M. Patterns of Damage in Genomic DNA Sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef]
- Glocke, I.; Meyer, M. Extending the Spectrum of DNA Sequences Retrieved from Ancient Bones and Teeth. Genome Res. 2017, 27, 1230–1237. [Google Scholar] [CrossRef]
- Damgaard, P.B.; Margaryan, A.; Schroeder, H.; Orlando, L.; Willerslev, E.; Allentoft, M.E. Improving Access to Endogenous DNA in Ancient Bones and Teeth. Sci. Rep. 2015, 5, 11184. [Google Scholar] [CrossRef]
- Korlević, P.; Gerber, T.; Gansauge, M.-T.; Hajdinjak, M.; Nagel, S.; Aximu-Petri, A.; Meyer, M. Reducing Microbial and Human Contamination in DNA Extractions from Ancient Bones and Teeth. Biotechniques 2015, 59, 87–93. [Google Scholar] [CrossRef]
- Gamba, C.; Hanghøj, K.; Gaunitz, C.; Alfarhan, A.H.; Alquraishi, S.A.; Al-Rasheid, K.A.S.; Bradley, D.G.; Orlando, L. Comparing the Performance of Three Ancient DNA Extraction Methods for High-throughput Sequencing. Mol. Ecol. Resour. 2016, 16, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Boessenkool, S.; Hanghøj, K.; Nistelberger, H.M.; Der Sarkissian, C.; Gondek, A.T.; Orlando, L.; Barrett, J.H.; Star, B. Combining Bleach and Mild Predigestion Improves Ancient DNA Recovery from Bones. Mol. Ecol. Resour. 2017, 17, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Nieves-Colón, M.A.; Ozga, A.T.; Pestle, W.J.; Cucina, A.; Tiesler, V.; Stanton, T.W.; Stone, A.C. Comparison of Two Ancient DNA Extraction Protocols for Skeletal Remains from Tropical Environments. Am. J. Phys. Anthropol. 2018, 166, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Salamon, M.; Tuross, N.; Arensburg, B.; Weiner, S. Relatively Well Preserved DNA Is Present in the Crystal Aggregates of Fossil Bones. Proc. Natl. Acad. Sci. USA 2005, 102, 13783–13788. [Google Scholar] [CrossRef]
- Campos, P.F.; Craig, O.E.; Turner-Walker, G.; Peacock, E.; Willerslev, E.; Gilbert, M.T.P. DNA in Ancient Bone–Where Is It Located and How Should We Extract It? Ann. Anat.-Anat. Anz. 2012, 194, 7–16. [Google Scholar] [CrossRef]
- Yang, D.Y.; Eng, B.; Waye, J.S.; Dudar, J.C.; Saunders, S.R. Improved DNA Extraction from Ancient Bones Using Silica-based Spin Columns. Am. J. Phys. Anthropol. Off. Publ. Am. Assoc. Phys. Anthropol. 1998, 105, 539–543. [Google Scholar] [CrossRef]
- Svensson, E.M.; Anderung, C.; Baubliene, J.; Persson, P.; Malmström, H.; Smith, C.; Vretemark, M.; Daugnora, L.; Götherström, A. Tracing Genetic Change over Time Using Nuclear SNPs in Ancient and Modern Cattle. Anim. Genet. 2007, 38, 378–383. [Google Scholar] [CrossRef]
- Anderung, C.; Persson, P.; Bouwman, A.; Elburg, R.; Götherström, A. Fishing for Ancient DNA. Forensic Sci. Int. Genet. 2008, 2, 104–107. [Google Scholar] [CrossRef]
- Ersmark, E.; Orlando, L.; Sandoval-Castellanos, E.; Barnes, I.; Barnett, R.; Stuart, A.; Lister, A.; Dalén, L. Population Demography and Genetic Diversity in the Pleistocene Cave Lion. Open Quat. 2015, 1, 1–14. [Google Scholar] [CrossRef]
- Krzewińska, M.; Kılınç, G.M.; Juras, A.; Koptekin, D.; Chyleński, M.; Nikitin, A.G.; Shcherbakov, N.; Shuteleva, I.; Leonova, T.; Kraeva, L. Ancient Genomes Suggest the Eastern Pontic-Caspian Steppe as the Source of Western Iron Age Nomads. Sci. Adv. 2018, 4, eaat4457. [Google Scholar] [CrossRef]
- Palkopoulou, E.; Dalen, L.; Lister, A.M.; Vartanyan, S.; Sablin, M.; Sher, A.; Edmark, V.N.; Brandstrom, M.D.; Germonpre, M.; Barnes, I.; et al. Holarctic Genetic Structure and Range Dynamics in the Woolly Mammoth. Proc. R. Soc. B Biol. Sci. 2013, 280, 20131910. [Google Scholar] [CrossRef]
- Leonard, J.A.; Wayne, R.K.; Cooper, A. From the Cover: Population Genetics of Ice Age Brown Bears. Proc. Natl. Acad. Sci. USA 2000, 97, 1651. [Google Scholar] [CrossRef]
- Meyer, M.; Kircher, M. Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and Sequencing. Cold Spring Harb. Protoc. 2010, 5. [Google Scholar] [CrossRef]
- Gansauge, M.-T.; Meyer, M. A Method for Single-Stranded Ancient DNA Library Preparation. In Ancient DNA; Humana Press: New York, NY, USA, 2019; pp. 75–83. [Google Scholar]
- Briggs, A.W.; Stenzel, U.; Meyer, M.; Krause, J.; Kircher, M.; Pääbo, S. Removal of Deaminated Cytosines and Detection of in Vivo Methylation in Ancient DNA. Nucleic Acids Res. 2010, 38, e87. [Google Scholar] [CrossRef]
- Der Sarkissian, C.; Ermini, L.; Schubert, M.; Yang, M.A.; Librado, P.; Fumagalli, M.; Jónsson, H.; Bar-Gal, G.K.; Albrechtsen, A.; Vieira, F.G.; et al. Evolutionary Genomics and Conservation of the Endangered Przewalski’s Horse. Curr. Biol. 2015, 25, 2577–2583. [Google Scholar] [CrossRef]
- Pečnerová, P.; Palkopoulou, E.; Wheat, C.W.; Skoglund, P.; Vartanyan, S.; Tikhonov, A.; Nikolskiy, P.; van der Plicht, J.; Díez-del-Molino, D.; Dalén, L. Mitogenome Evolution in the Last Surviving Woolly Mammoth Population Reveals Neutral and Functional Consequences of Small Population Size. Evol. Lett. 2017, 1, 292–303. [Google Scholar] [CrossRef]
- Rohland, N.; Harney, E.; Mallick, S.; Nordenfelt, S.; Reich, D. Partial Uracil–DNA–Glycosylase Treatment for Screening of Ancient DNA. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20130624. [Google Scholar] [CrossRef]
- Adler, C.J.; Haak, W.; Donlon, D.; Cooper, A.; Consortium, G. Survival and Recovery of DNA from Ancient Teeth and Bones. J. Archaeol. Sci. 2011, 38, 956–964. [Google Scholar] [CrossRef]
- Daley, T.; Smith, A.D. Predicting the Molecular Complexity of Sequencing Libraries. Nat. Methods 2013, 10, 325. [Google Scholar] [CrossRef]
- Kutschera, V.E.; Kierczak, M.; van der Valk, T.; von Seth, J.; Dussex, N.; Lord, E.; Dehasque, M.; Stanton, D.W.G.; Khoonsari, P.E.; Nystedt, B.; et al. GenErode: A Bioinformatics Pipeline to Investigate Genome Erosion in Endangered and Extinct Species. bioRxiv 2022. [Google Scholar] [CrossRef]
- Köster, J.; Rahmann, S. Snakemake—A Scalable Bioinformatics Workflow Engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef]
- Krause, J.; Dear, P.H.; Pollack, J.L.; Slatkin, M.; Spriggs, H.; Barnes, I.; Lister, A.M.; Ebersberger, I.; Pääbo, S.; Hofreiter, M. Multiplex Amplification of the Mammoth Mitochondrial Genome and the Evolution of Elephantidae. Nature 2006, 439, 724. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.F.; Orlando, L. MapDamage2. 0: Fast Approximate Bayesian Estimates of Ancient DNA Damage Parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef]
- Feuerborn, T.R.; Palkopoulou, E.; van der Valk, T.; von Seth, J.; Munters, A.R.; Pečnerová, P.; Dehasque, M.; Ureña, I.; Ersmark, E.; Lagerholm, V.K.; et al. Competitive Mapping Allows for the Identification and Exclusion of Human DNA Contamination in Ancient Faunal Genomic Datasets. BMC Genom. 2020, 21, 844. [Google Scholar] [CrossRef]
- Malmström, H.; Svensson, E.M.; Gilbert, M.T.P.; Willerslev, E.; Götherström, A.; Holmlund, G. More on Contamination: The Use of Asymmetric Molecular Behavior to Identify Authentic Ancient Human DNA. Mol. Biol. Evol. 2007, 24, 998–1004. [Google Scholar] [CrossRef]
- Basler, N.; Xenikoudakis, G.; Westbury, M.V.; Song, L.; Sheng, G.; Barlow, A. Reduction of the Contaminant Fraction of DNA Obtained from an Ancient Giant Panda Bone. BMC Res. Notes 2017, 10, 754. [Google Scholar] [CrossRef]
- Pečnerová, P.; Díez-del-Molino, D.; Dussex, N.; Feuerborn, T.; von Seth, J.; van der Plicht, J.; Nikolskiy, P.; Tikhonov, A.; Vartanyan, S.; Dalén, L. Genome-Based Sexing Provides Clues about Behavior and Social Structure in the Woolly Mammoth. Curr. Biol. 2017, 27, 3505–3510.e3. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcera, M.; Gigli, E.; Sanchez-Quinto, F.; Ramirez, O.; Calafell, F.; Civit, S.; Lalueza-Fox, C. Fragmentation of Contaminant and Endogenous DNA in Ancient Samples Determined by Shotgun Sequencing; Prospects for Human Palaeogenomics. PLoS ONE 2011, 6, e24161. [Google Scholar] [CrossRef] [PubMed]
- Varshney, U.; van de Sande, J.H. Specificities and Kinetics of Uracil Excision from Uracil-Containing DNA Oligomers by Escherichia Coli Uracil DNA Glycosylase. Biochemistry 1991, 30, 4055–4061. [Google Scholar] [CrossRef] [PubMed]
- Renaud, G.; Schubert, M.; Sawyer, S.; Orlando, L. Authentication and Assessment of Contamination in Ancient DNA. In Ancient DNA; Humana Press: New York, NY, USA, 2019; pp. 163–194. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Paired Test | Test Statistic | p Value | ||
|---|---|---|---|---|
| Predigestion (n = 6) | Complexity | t-test | t = 4.061 | 0.00486 |
| Endogenous content | t-test | t = −3.135 | 0.01291 | |
| Average fragment length | t-test | t = 2.019 | 0.04974 | |
| GC content | Wilcoxon signed ranks | V = 9.5 | 0.6241 | |
| Coverage | t-test | t = −1.562 | 0.08947 | |
| Damage parameter δD | t-test | t = 1.633 | 0.08168 | |
| Damage parameter δS | t-test | t = −0.009815 | 0.4963 | |
| Damage parameter λ | t-test | t = 1.765 | 0.06888 | |
| Bleach wash and Predigestion (n = 7) | Complexity | t-test | t = 5.156 | 0.001052 |
| Endogenous content | t-test | t = −1.748 | 0.06555 | |
| Average fragment length | t-test | t = −0.1617 | 0.4384 | |
| GC content | t-test | t = 4.514 | 0.002023 | |
| Coverage | t-test | t = −1.327 | 0.1164 | |
| Damage parameter δD | t-test | t = −4.912 | 0.00134 | |
| Damage parameter δD | t-test | t = −8.278 | 0.00008414 | |
| Damage parameter λ | t-test | t = −3.797 | 0.004502 | |
| MinElute column (n = 5) | Endogenous content | Wilcoxon signed ranks | V = 7 | 0.5 |
| Complexity | t-test | t = −0.615 | 0.2859 | |
| Average fragment length | t-test | t = −2.3524 | 0.03916 | |
| GC content | Wilcoxon signed ranks | V = 15 | 0.03125 | |
| Coverage | Wilcoxon signed ranks | V = 5 | 0.3125 | |
| Damage parameter δD | Wilcoxon signed ranks | V = 7 | 0.5 | |
| Damage parameter δS | Wilcoxon signed ranks | V = 12 | 0.1562 | |
| Damage parameter λ | Wilcoxon signed ranks | V = 15 | 0.03125 | |
| 10 MWCO filter (n = 7) | Endogenous content | t-test | t = −1.815 | 0.0597 |
| Complexity | t-test | t = −0.06492 | 0.47515 | |
| Average fragment length | t-test | t = 2.002 | 0.0461 | |
| GC content | Wilcoxon signed ranks | V = 1 | 0.5 | |
| Coverage | Wilcoxon signed ranks | V = 18 | 0.28905 | |
| Damage parameter δD | Wilcoxon signed ranks | V = 18 | 0.28905 | |
| Damage parameter δS | Wilcoxon signed ranks | V = 25 | 0.03906 | |
| Damage parameter λ | Wilcoxon signed ranks | V = 18 | 0.28905 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dehasque, M.; Pečnerová, P.; Kempe Lagerholm, V.; Ersmark, E.; Danilov, G.K.; Mortensen, P.; Vartanyan, S.; Dalén, L. Development and Optimization of a Silica Column-Based Extraction Protocol for Ancient DNA. Genes 2022, 13, 687. https://doi.org/10.3390/genes13040687
Dehasque M, Pečnerová P, Kempe Lagerholm V, Ersmark E, Danilov GK, Mortensen P, Vartanyan S, Dalén L. Development and Optimization of a Silica Column-Based Extraction Protocol for Ancient DNA. Genes. 2022; 13(4):687. https://doi.org/10.3390/genes13040687
Chicago/Turabian StyleDehasque, Marianne, Patrícia Pečnerová, Vendela Kempe Lagerholm, Erik Ersmark, Gleb K. Danilov, Peter Mortensen, Sergey Vartanyan, and Love Dalén. 2022. "Development and Optimization of a Silica Column-Based Extraction Protocol for Ancient DNA" Genes 13, no. 4: 687. https://doi.org/10.3390/genes13040687
APA StyleDehasque, M., Pečnerová, P., Kempe Lagerholm, V., Ersmark, E., Danilov, G. K., Mortensen, P., Vartanyan, S., & Dalén, L. (2022). Development and Optimization of a Silica Column-Based Extraction Protocol for Ancient DNA. Genes, 13(4), 687. https://doi.org/10.3390/genes13040687