
Comparative Analysis of Transposable Elements and the Identification of Candidate Centromeric Elements in the Prunus Subgenus Cerasus and Its Relatives

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Dataset and Plant Material
2.2. TE Identification, Classification and Annotation
2.3. Full-Length Long Terminal Repeat Retrotransposon Identification
2.4. Tandem Repeats and Centromere Prediction
2.5. Chromosome Preparation, Probe Synthesis and FISH
2.6. Identification and Phylogenetic Analysis of Centromeric Histone H3
3. Results
3.1. Comparative Analysis of Transposable Elements
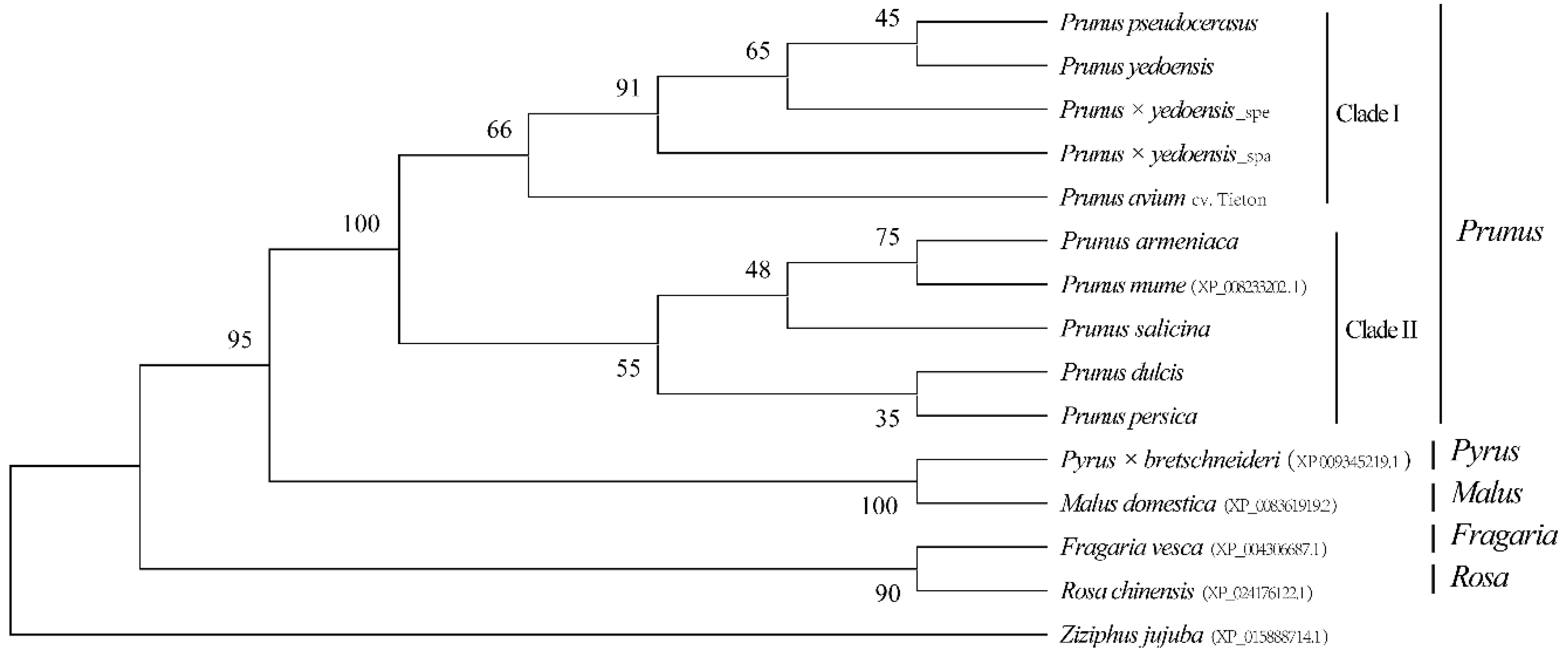
3.2. Characterization and Similarity of LTR-RT Sequences
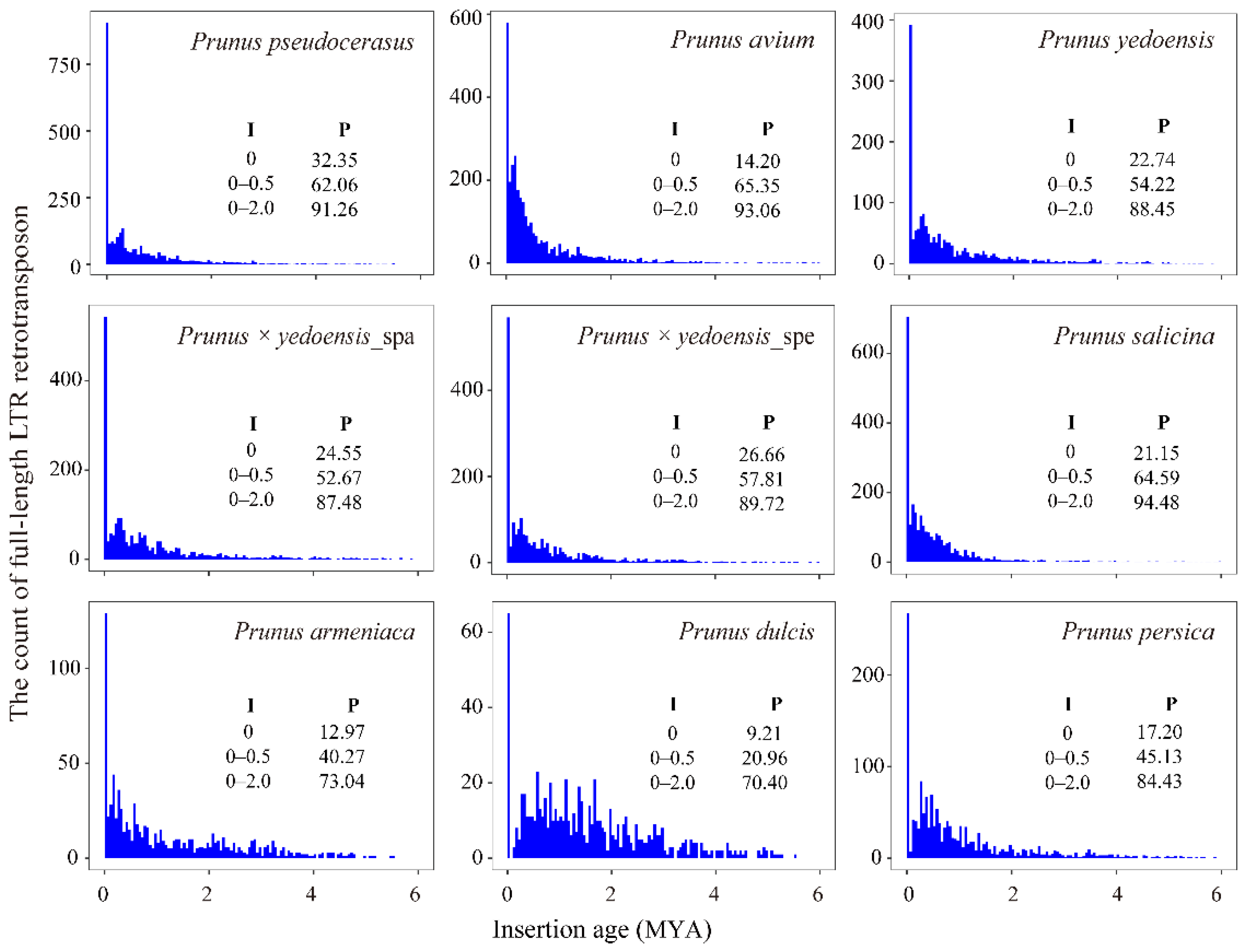
3.3. LTR Insertion Time Estimation
3.4. Characterization of Tandem Repeats
3.5. Centromeric and Telomeric Distribution on Chromosomes
3.6. Conservation of Centromere-Specific Histone H3
4. Discussion
4.1. Genome Size Expansion through the Amplification of Repetitive Sequences
4.2. LTR-RTs Drive Genome Evolution
4.3. Conservation of the Centromere Sequence and CENH3
4.4. FISH Is a Method for Directly Correcting Assembled Genomes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riva, S.C.; Opara, U.O.; Fawole, O.A. Recent developments on postharvest application of edible coatings on stone fruit: A review. Sci. Hortic. 2020, 262, 109074. [Google Scholar] [CrossRef]
- Baek, S.; Choi, K.; Kim, G.B.; Yu, H.J.; Cho, A.; Jang, H.; Kim, C.; Kim, H.J.; Chang, K.S.; Kim, J.H.; et al. Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries. Genome Biol. 2018, 19, 127. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.-G.; Yu, X.-Q.; Chen, J.; Zhang, M.; Liu, S.-W.; Zhu, H.; Li, M.; Duan, Y.-F.; Chen, L.; Wu, L.; et al. The genome of Chinese flowering cherry (Cerasus serrulata) provides new insights into Cerasus species. Hortic. Res. 2020, 7, 165. [Google Scholar] [CrossRef] [PubMed]
- Tetreault, H.M.; Ungerer, M.C. Long Terminal Repeat Retrotransposon Content in Eight Diploid Sunflower Species Inferred from Next-Generation Sequence Data. G3 2016, 6, 2299–2308. [Google Scholar] [CrossRef]
- Mascagni, F.; Vangelisti, A.; Giordani, T.; Cavallini, A.; Natali, L. Specific LTR-Retrotransposons Show Copy Number Variations between Wild and Cultivated Sunflowers. Genes 2018, 9, 433. [Google Scholar] [CrossRef]
- Vitales, D.; Garcia, S.; Dodsworth, S. Reconstructing phylogenetic relationships based on repeat sequence similarities. Mol. Phylogenet. Evol. 2020, 147, 106766. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu Rev. Plant Biol. 2014, 65, 505–530. [Google Scholar] [CrossRef]
- Zavallo, D.; Crescente, J.M.; Gantuz, M.; Leone, M.; Vanzetti, L.S.; Masuelli, R.W.; Asurmendi, S. Genomic re-assessment of the transposable element landscape of the potato genome. Plant Cell Rep. 2020, 39, 1161–1174. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, J.; Han, X.; Li, J.; Gao, Y.; Richards, C.M.; Zhang, C.; Tian, Y.; Liu, G.; Gul, H.; et al. A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 2019, 10, 1494. [Google Scholar] [CrossRef]
- McCann, J.; Macas, J.; Novak, P.; Stuessy, T.F.; Villasenor, J.L.; Weiss-Schneeweiss, H. Differential Genome Size and Repetitive DNA Evolution in Diploid Species of Melampodium sect. Melampodium (Asteraceae). Front. Plant Sci. 2020, 11, 362. [Google Scholar] [CrossRef]
- Zeng, X.; Xu, T.; Ling, Z.; Wang, Y.; Li, X.; Xu, S.; Xu, Q.; Zha, S.; Qimei, W.; Basang, Y.; et al. An improved high-quality genome assembly and annotation of Tibetan hulless barley. Sci. Data 2020, 7, 139. [Google Scholar] [CrossRef] [PubMed]
- Butelli, E.; Licciardello, C.; Zhang, Y.; Liu, J.; Mackay, S.; Bailey, P.; Reforgiato-Recupero, G.; Martin, C. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 2012, 24, 1242–1255. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, S.; Goyal, V. Repetitive sequences in plant nuclear DNA: Types, distribution, evolution and function. Genom. Proteom. Bioinform. 2014, 12, 164–171. [Google Scholar] [CrossRef]
- Li, S.F.; Su, T.; Cheng, G.Q.; Wang, B.X.; Li, X.; Deng, C.L.; Gao, W.J. Chromosome Evolution in Connection with Repetitive Sequences and Epigenetics in Plants. Genes 2017, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J. Fluorescence in situ hybridization in plants: Recent developments and future applications. Chromosome Res. 2019, 27, 153–165. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, W.; Sun, L.; Zhao, F.; Huang, B.; Yang, W.; Tao, Y.; Wang, J.; Yuan, Z.; Fan, G.; et al. The genome of Prunus mume. Nat. Commun. 2012, 3, 1318. [Google Scholar] [CrossRef]
- The International Peach Genome Initiative; Verde, I.; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef]
- Verde, I.; Jenkins, J.; Dondini, L.; Micali, S.; Pagliarani, G.; Vendramin, E.; Paris, R.; Aramini, V.; Gazza, L.; Rossini, L.; et al. The Peach v2.0 release: High-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Genom. 2017, 18, 225. [Google Scholar] [CrossRef]
- Tan, Q.; Li, S.; Zhang, Y.; Chen, M.; Wen, B.; Jiang, S.; Chen, X.; Fu, X.; Li, D.; Wu, H.; et al. Chromosome-level genome assemblies of five Prunus species and genome-wide association studies for key agronomic traits in peach. Hortic. Res. 2021, 8, 213. [Google Scholar] [CrossRef]
- Shirasawa, K.; Isuzugawa, K.; Ikenaga, M.; Saito, Y.; Yamamoto, T.; Hirakawa, H.; Isobe, S. The genome sequence of sweet cherry (Prunus avium) for use in genomics-assisted breeding. DNA Res. 2017, 24, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Pinosio, S.; Marroni, F.; Zuccolo, A.; Vitulo, N.; Mariette, S.; Sonnante, G.; Aravanopoulos, F.A.; Ganopoulos, I.; Palasciano, M.; Vidotto, M.; et al. A draft genome of sweet cherry (Prunus avium L.) reveals genome-wide and local effects of domestication. Plant J. 2020, 103, 1420–1432. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, W.; Zhu, D.; Hong, P.; Zhang, S.; Xiao, S.; Tan, Y.; Chen, X.; Xu, L.; Zong, X.; et al. Chromosome-scale genome assembly of sweet cherry (Prunus avium L.) cv. Tieton obtained using long-read and Hi-C sequencing. Hortic. Res. 2020, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, K.; Esumi, T.; Hirakawa, H.; Tanaka, H.; Itai, A.; Ghelfi, A.; Nagasaki, H.; Isobe, S. Phased genome sequence of an interspecific hybrid flowering cherry, ’Somei-Yoshino’ (Cerasus × yedoensis). DNA Res. 2019, 26, 379–389. [Google Scholar] [CrossRef]
- Shirasawa, K.; Itai, A.; Isobe, S. Genome sequencing and analysis of two early-flowering cherry (Cerasus × kanzakura) varieties, ‘Kawazu-zakura’ and ‘Atami-zakura’. DNA Res. 2021, 28, dsab026. [Google Scholar] [CrossRef]
- Sanchez-Perez, R.; Pavan, S.; Mazzeo, R.; Moldovan, C.; Aiese Cigliano, R.; Del Cueto, J.; Ricciardi, F.; Lotti, C.; Ricciardi, L.; Dicenta, F.; et al. Mutation of a bHLH transcription factor allowed almond domestication. Science 2019, 364, 1095–1098. [Google Scholar] [CrossRef]
- Alioto, T.; Alexiou, K.G.; Bardil, A.; Barteri, F.; Castanera, R.; Cruz, F.; Dhingra, A.; Duval, H.; Fernandez, I.M.A.; Frias, L.; et al. Transposons played a major role in the diversification between the closely related almond and peach genomes: Results from the almond genome sequence. Plant J. 2020, 101, 455–472. [Google Scholar] [CrossRef]
- Jiang, F.; Zhang, J.; Wang, S.; Yang, L.; Luo, Y.; Gao, S.; Zhang, M.; Wu, S.; Hu, S.; Sun, H.; et al. The apricot (Prunus armeniaca L.) genome elucidates Rosaceae evolution and beta-carotenoid synthesis. Hortic. Res. 2019, 6, 128. [Google Scholar] [CrossRef]
- Liu, C.; Feng, C.; Peng, W.; Hao, J.; Wang, J.; Pan, J.; He, Y. Chromosome-level draft genome of a diploid plum (Prunus salicina). Gigascience 2020, 9, giaa130. [Google Scholar] [CrossRef]
- Huang, Z.; Shen, F.; Chen, Y.; Cao, K.; Wang, L. Chromosome-scale genome assembly and population genomics provide insights into the adaptation, domestication, and flavonoid metabolism of Chinese plum. Plant J. 2021, 108, 1174–1192. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Bombarely, A.; Li, S. DeepTE: A computational method for de novo classification of transposons with convolutional neural network. Bioinformatics 2020, 36, 4269–4275. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef]
- Valencia, J.D.; Girgis, H.Z. LtrDetector: A tool-suite for detecting long terminal repeat retrotransposons de-novo. BMC Genom. 2019, 20, 450. [Google Scholar] [CrossRef]
- Ou, S.; Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef]
- Xie, Z.; Wang, L.; Wang, L.; Wang, Z.; Lu, Z.; Tian, D.; Yang, S.; Hurst, L.D. Mutation rate analysis via parent-progeny sequencing of the perennial peach. I. A low rate in woody perennials and a higher mutagenicity in hybrids. Proc. Biol. Sci. 2016, 283, 20161016. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Sobreira, T.J.; Durham, A.M.; Gruber, A. TRAP: Automated classification, quantification and annotation of tandemly repeated sequences. Bioinformatics 2006, 22, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Neumann, P.; Macas, J. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat. Protoc. 2020, 15, 3745–3776. [Google Scholar] [CrossRef] [PubMed]
- Melters, D.P.; Bradnam, K.R.; Young, H.A.; Telis, N.; May, M.R.; Ruby, J.G.; Sebra, R.; Peluso, P.; Eid, J.; Rank, D.; et al. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 2013, 14, R10. [Google Scholar] [CrossRef] [PubMed]
- Brodie, R.; Roper, R.L.; Upton, C. JDotter: A Java interface to multiple dotplots generated by dotter. Bioinformatics 2004, 20, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Chen, Q.; Zhang, L.; Tang, H.; Luo, Y.; Liu, Z. Phylogenetic insight into subgenera Idaeobatus and Malachobatus (Rubus, Rosaceae) inferring from ISH analysis. Mol. Cytogenet. 2015, 8, 11. [Google Scholar] [CrossRef]
- Du, P.; Li, L.; Liu, H.; Fu, L.; Qin, L.; Zhang, Z.; Cui, C.; Sun, Z.; Han, S.; Xu, J.; et al. High-resolution chromosome painting with repetitive and single-copy oligonucleotides in Arachis species identifies structural rearrangements and genome differentiation. BMC Plant Biol. 2018, 18, 240. [Google Scholar] [CrossRef]
- Hlouskova, P.; Mandakova, T.; Pouch, M.; Travnicek, P.; Lysak, M.A. The large genome size variation in the Hesperis clade was shaped by the prevalent proliferation of DNA repeats and rarer genome downsizing. Ann. Bot. 2019, 124, 103–120. [Google Scholar] [CrossRef]
- Ichiyanagi, K.; Saito, K. TE studies in Japan: The fourth Japanese meeting on host-transposon interactions. Mob. DNA 2019, 10, 11. [Google Scholar] [CrossRef]
- Balzano, E.; Pelliccia, F.; Giunta, S. Genome (in) stability at tandem repeats. Semin. Cell Dev. Biol. 2021, 113, 97–112. [Google Scholar] [CrossRef]
- Alix, K.; Gerard, P.R.; Schwarzacher, T.; Heslop-Harrison, J.S.P. Polyploidy and interspecific hybridization: Partners for adaptation, speciation and evolution in plants. Ann. Bot. 2017, 120, 183–194. [Google Scholar] [CrossRef]
- Vicient, C.M.; Casacuberta, J.M. Impact of transposable elements on polyploid plant genomes. Ann. Bot. 2017, 120, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Vitales, D.; Alvarez, I.; Garcia, S.; Hidalgo, O.; Nieto Feliner, G.; Pellicer, J.; Valles, J.; Garnatje, T. Genome size variation at constant chromosome number is not correlated with repetitive DNA dynamism in Anacyclus (Asteraceae). Ann. Bot. 2020, 125, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Stritt, C.; Wyler, M.; Gimmi, E.L.; Pippel, M.; Roulin, A.C. Diversity, dynamics and effects of long terminal repeat retrotransposons in the model grass Brachypodium distachyon. New Phytol. 2020, 227, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jiao, C.; Schwaninger, H.; Chao, C.T.; Ma, Y.; Duan, N.; Khan, A.; Ban, S.; Xu, K.; Cheng, L.; et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat. Genet. 2020, 52, 1423–1432. [Google Scholar] [CrossRef]
- Zhang, J.; Lei, Y.; Wang, B.; Li, S.; Yu, S.; Wang, Y.; Li, H.; Liu, Y.; Ma, Y.; Dai, H.; et al. The high-quality genome of diploid strawberry (Fragaria nilgerrensis) provides new insights into anthocyanin accumulation. Plant Biotechnol. J. 2020, 18, 1908–1924. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, Y.; Liu, F.; Zhang, S.; Wang, X.; Lu, Q.; Wang, K.; Zhang, B.; Peng, R. Genome-Wide Survey and Comparative Analysis of Long Terminal Repeat (LTR) Retrotransposon Families in Four Gossypium Species. Sci. Rep. 2018, 8, 9399. [Google Scholar] [CrossRef]
- de Assis, R.; Baba, V.Y.; Cintra, L.A.; Goncalves, L.S.A.; Rodrigues, R.; Vanzela, A.L.L. Genome relationships and LTR-retrotransposon diversity in three cultivated Capsicum L. (Solanaceae) species. BMC Genom. 2020, 21, 237. [Google Scholar] [CrossRef]
- Han, M.; Yang, Y.; Zhang, M.; Wang, K. Considerations regarding centromere assembly in plant whole-genome sequencing. Methods 2021, 187, 54–56. [Google Scholar] [CrossRef]
- Comai, L.; Maheshwari, S.; Marimuthu, M.P.A. Plant centromeres. Curr. Opin. Plant Biol. 2017, 36, 158–167. [Google Scholar] [CrossRef]
- He, Q.; Cai, Z.; Hu, T.; Liu, H.; Bao, C.; Mao, W.; Jin, W. Repetitive sequence analysis and karyotyping reveals centromere-associated DNA sequences in radish (Raphanus sativus L.). BMC Plant Biol. 2015, 15, 105. [Google Scholar] [CrossRef]
- Iwata-Otsubo, A.; Radke, B.; Findley, S.; Abernathy, B.; Vallejos, C.E.; Jackson, S.A. Fluorescence In Situ Hybridization (FISH)-Based Karyotyping Reveals Rapid Evolution of Centromeric and Subtelomeric Repeats in Common Bean (Phaseolus vulgaris) and Relatives. G3 2016, 6, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Hibrand Saint-Oyant, L.; Ruttink, T.; Hamama, L.; Kirov, I.; Lakhwani, D.; Zhou, N.N.; Bourke, P.M.; Daccord, N.; Leus, L.; Schulz, D.; et al. A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat. Plants 2018, 4, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Lunerova, J.; Herklotz, V.; Laudien, M.; Vozarova, R.; Groth, M.; Kovarik, A.; Ritz, C.M. Asymmetrical canina meiosis is accompanied by the expansion of a pericentromeric satellite in non-recombining univalent chromosomes in the genus Rosa. Ann. Bot. 2020, 125, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhao, H.; Zhang, T.; Zeng, Z.; Zhang, P.; Zhu, B.; Han, Y.; Braz, G.T.; Casler, M.D.; Schmutz, J.; et al. Amplification and adaptation of centromeric repeats in polyploid switchgrass species. New Phytol. 2018, 218, 1645–1657. [Google Scholar] [CrossRef]
- VanBuren, R.; Wai, C.M.; Colle, M.; Wang, J.; Sullivan, S.; Bushakra, J.M.; Liachko, I.; Vining, K.J.; Dossett, M.; Finn, C.E.; et al. A near complete, chromosome-scale assembly of the black raspberry (Rubus occidentalis) genome. Gigascience 2018, 7, giy094. [Google Scholar] [CrossRef]
- Bureš, P.; Zedek, F. Holokinetic drive: Centromere drive in chromosomes without centromeres. Evolution 2014, 68, 2412–2420. [Google Scholar] [CrossRef][Green Version]
- Hodel, R.G.J.; Zimmer, E.; Wen, J. A phylogenomic approach resolves the backbone of Prunus (Rosaceae) and identifies signals of hybridization and allopolyploidy. Mol. Phylogenet. Evol. 2021, 160, 107118. [Google Scholar] [CrossRef]
- Zhang, S.D.; Jin, J.J.; Chen, S.Y.; Chase, M.W.; Soltis, D.E.; Li, H.T.; Yang, J.B.; Li, D.Z.; Yi, T.S. Diversification of Rosaceae since the Late Cretaceous based on plastid phylogenomics. New Phytol. 2017, 214, 1355–1367. [Google Scholar] [CrossRef]
- Xiang, Y.; Huang, C.H.; Hu, Y.; Wen, J.; Li, S.; Yi, T.; Chen, H.; Xiang, J.; Ma, H. Evolution of Rosaceae Fruit Types Based on Nuclear Phylogeny in the Context of Geological Times and Genome Duplication. Mol. Biol. Evol. 2017, 34, 262–281. [Google Scholar] [CrossRef]
- Nagaki, K.; Tanaka, K.; Yamaji, N.; Kobayashi, H.; Murata, M. Sunflower centromeres consist of a centromere-specific LINE and a chromosome-specific tandem repeat. Front. Plant Sci. 2015, 6, 912. [Google Scholar] [CrossRef]
- Ishii, T.; Juranic, M.; Maheshwari, S.; Bustamante, F.O.; Vogt, M.; Salinas-Gamboa, R.; Dreissig, S.; Gursanscky, N.; How, T.; Demidov, D.; et al. Unequal contribution of two paralogous CENH3 variants in cowpea centromere function. Commun. Biol. 2020, 3, 775. [Google Scholar] [CrossRef] [PubMed]
- Evtushenko, E.V.; Elisafenko, E.A.; Gatzkaya, S.S.; Lipikhina, Y.A.; Houben, A.; Vershinin, A.V. Conserved molecular structure of the centromeric histone CENH3 in Secale and its phylogenetic relationships. Sci. Rep. 2017, 7, 17628. [Google Scholar] [CrossRef] [PubMed]
- Kratka, M.; Smerda, J.; Lojdova, K.; Bures, P.; Zedek, F. Holocentric Chromosomes Probably Do Not Prevent Centromere Drive in Cyperaceae. Front. Plant Sci. 2021, 12, 642661. [Google Scholar] [CrossRef] [PubMed]
- Lou, Q.; Zhang, Y.; He, Y.; Li, J.; Jia, L.; Cheng, C.; Guan, W.; Yang, S.; Chen, J. Single-copy gene-based chromosome painting in cucumber and its application for chromosome rearrangement analysis in Cucumis. Plant J. 2014, 78, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Koo, D.H.; Li, Y.; Zhang, X.; Luan, F.; Havey, M.J.; Jiang, J.; Weng, Y. Chromosome rearrangements during domestication of cucumber as revealed by high-density genetic mapping and draft genome assembly. Plant J. 2012, 71, 895–906. [Google Scholar] [CrossRef]
- Karafiatova, M.; Bartos, J.; Kopecky, D.; Ma, L.; Sato, K.; Houben, A.; Stein, N.; Dolezel, J. Mapping nonrecombining regions in barley using multicolor FISH. Chromosome Res. 2013, 21, 739–751. [Google Scholar] [CrossRef]
- Shearer, L.A.; Anderson, L.K.; de Jong, H.; Smit, S.; Goicoechea, J.L.; Roe, B.A.; Hua, A.; Giovannoni, J.J.; Stack, S.M. Fluorescence in situ hybridization and optical mapping to correct scaffold arrangement in the tomato genome. G3 2014, 4, 1395–1405. [Google Scholar] [CrossRef]
- Meng, Z.; Hu, X.; Zhang, Z.; Li, Z.; Lin, Q.; Yang, M.; Yang, P.; Ming, R.; Yu, Q.; Wang, K. Chromosome Nomenclature and Cytological Characterization of Sacred Lotus. Cytogenet. Genome Res. 2017, 153, 223–231. [Google Scholar] [CrossRef]
- Tang, L. Genomics beyond complete genomes. Nat. Methods 2022, 19, 29. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | Reference Genomes | Origin and Chromosome Number of Samples Used in Molecular Cytogenetic Analyses | ||
|---|---|---|---|---|
| Code | Origin | Ploidy Level | ||
| subg. Cerasus | ||||
| Prunus pseudocerasus | ‘Luoyang Guying’ (unpublished) | HC | Miyi, Sichuan, China | 2n = 4x = 32 |
| XC1 | Xichang, Sichuan, China | |||
| Prunus avium | ‘Tieton’ (v2.0) [23] | Van | ZFI, CAAS, China | 2n = 2x = 16 |
| Mazzard | ||||
| Prunus yedoensis | ‘Pyn-Jeju2′ (v1.0) [2] | Pyedoensis | Chengdu, Sichuan, China | 2n = 2x = 16 |
| Prunus × yedoensis | ‘Somei-yoshino’ a (v3.1) [24] | - | - | 2n = 2x = 16 |
| Prunus campanulata | - | Pcampan | Chengdu, Sichuan, China | 2n = 2x = 16 |
| subg. Prunus | ||||
| Prunus tomentosa | - | red_fruit | ZFI, CAAS, China | 2n = 2x = 16 |
| white_fruit | 2n = 2x = 16 | |||
| Prunus humilis | - | Phumilis | Suqian, Jiangsu, China | 2n = 2x = 16 |
| Prunus salicina | ‘Sanyueli’ (v1.0) [29] | Cuihongli | Chengdu, Sichuan, China | 2n = 2x = 16 |
| subg. Armeniaca | ||||
| Prunus armeniaca | ‘Chuanzhihong’ (v1.0) [28] | Diaogan | Akesu, Xinjiang, China | 2n = 2x = 16 |
| subg. Amygdalus | ||||
| Prunusdulcis | ‘Texas’ (v2.0) [27] | Pdulcis | Luntai, Xinjiang, China | 2n = 2x = 16 |
| Prunus persica | ‘Lovell’ (v2.0) [19] | Ppersica | Chengdu, Sichuan, China | 2n = 2x = 16 |
| Genome | Prunus pseudocerasus | Prunus avium | Prunus yedoensis | Cerasus × yedoensis (spa/spe) | Prunus salicina | Prunus armeniaca | Prunus dulcis | Prunus persica |
|---|---|---|---|---|---|---|---|---|
| LINE | 1.87 | 1.56 | 1.88 | 1.75/1.81 | 1.69 | 1.71 | 1.51 | 1.5 |
| SINE | 0.3 | 0.23 | 0.31 | 0.3/0.3 | 0.29 | 0.33 | 0.3 | 0.29 |
| LTR | 27.66 | 29.03 | 24.01 | 24.87/23.79 | 31.38 | 21.16 | 24.83 | 23.23 |
| Copia | 10.76 | 8.19 | 9.5 | 9.73/9.11 | 9.32 | 7.48 | 9.06 | 9.43 |
| Gypsy | 16.9 | 20.84 | 14.51 | 15.14/14.68 | 22.06 | 13.68 | 15.77 | 13.8 |
| nLTR | 0.21 | 0.17 | 0.2 | 0.23/0.23 | 0.19 | 0.24 | 0.16 | 0.16 |
| DIRS | 0.03 | 0.03 | 0.03 | 0.03/0.03 | 0.03 | 0.03 | 0.02 | 0.02 |
| PLE | 0.18 | 0.14 | 0.17 | 0.2/0.2 | 0.16 | 0.21 | 0.14 | 0.14 |
| Subclass_1 | 27.82 | 22.06 | 25.76 | 26.93/27.91 | 20.77 | 24.97 | 22.94 | 25.61 |
| TIR/CACTA | 8.12 | 4.5 | 6.58 | 6.58/7.03 | 4.25 | 4.43 | 5.07 | 7.41 |
| TIR/MuDR | 7.9 | 5.68 | 7.83 | 7.93/7.78 | 6.1 | 8.14 | 7.16 | 7.56 |
| TIR/PIF-Harbinger | 3.25 | 5.56 | 3.24 | 3.95/5.14 | 3.06 | 3.73 | 3.65 | 3.47 |
| TIR/Tc1-Mariner | 2.73 | 2.12 | 2.69 | 2.77/2.6 | 2.37 | 2.82 | 2.3 | 2.38 |
| TIR/hAT | 5.82 | 4.2 | 5.42 | 5.7/5.36 | 4.99 | 5.85 | 4.76 | 4.79 |
| Subclass_2 | 4.01 | 2.77 | 3.98 | 3.95/3.93 | 3.71 | 3.85 | 3.56 | 3.63 |
| Helitron | 3.37 | 2.26 | 3.31 | 3.3/3.3 | 3.12 | 3.22 | 2.95 | 3.04 |
| MITE | 0.64 | 0.51 | 0.67 | 0.65/0.63 | 0.59 | 0.63 | 0.61 | 0.59 |
| Total | 61.86 | 55.81 | 56.14 | 58.03/57.94 | 58.05 | 52.28 | 53.31 | 54.41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Wang, Y.; Zhang, J.; Feng, Y.; Chen, Q.; Liu, Z.-S.; Liu, C.-L.; He, W.; Wang, H.; Yang, S.-F.; et al. Comparative Analysis of Transposable Elements and the Identification of Candidate Centromeric Elements in the Prunus Subgenus Cerasus and Its Relatives. Genes 2022, 13, 641. https://doi.org/10.3390/genes13040641
Wang L, Wang Y, Zhang J, Feng Y, Chen Q, Liu Z-S, Liu C-L, He W, Wang H, Yang S-F, et al. Comparative Analysis of Transposable Elements and the Identification of Candidate Centromeric Elements in the Prunus Subgenus Cerasus and Its Relatives. Genes. 2022; 13(4):641. https://doi.org/10.3390/genes13040641
Chicago/Turabian StyleWang, Lei, Yan Wang, Jing Zhang, Yan Feng, Qing Chen, Zhen-Shan Liu, Cong-Li Liu, Wen He, Hao Wang, Shao-Feng Yang, and et al. 2022. "Comparative Analysis of Transposable Elements and the Identification of Candidate Centromeric Elements in the Prunus Subgenus Cerasus and Its Relatives" Genes 13, no. 4: 641. https://doi.org/10.3390/genes13040641
APA StyleWang, L., Wang, Y., Zhang, J., Feng, Y., Chen, Q., Liu, Z.-S., Liu, C.-L., He, W., Wang, H., Yang, S.-F., Zhang, Y., Luo, Y., Tang, H.-R., & Wang, X.-R. (2022). Comparative Analysis of Transposable Elements and the Identification of Candidate Centromeric Elements in the Prunus Subgenus Cerasus and Its Relatives. Genes, 13(4), 641. https://doi.org/10.3390/genes13040641