
The Complete Mitochondrial Genome of the Chicken Body Louse, Menacanthus cornutus, and Evolutionary Patterns of Extensive Gene Rearrangements in the Mitochondrial Genomes of Amblycera (Psocodea: Phthiraptera)




Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, Genomic DNA Extraction, and Sequence Amplification
2.2. Mitochondrial Genome Sequencing, Assembly, and Annotation
2.3. Phylogenetic Analyses
3. Results and Discussion
3.1. The General Features of the Menacanthus cornutus mt Genome
3.2. Phylogenetic Analyses of the Suborder Amblycera and Other Parasitic Lice
3.3. Evolution of Gene Rearrangement in the Mitochondrial Genome of Amblycera
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wallace, D.C. Structure and evolution of organelle genomes. Microbiol. Rev. 1982, 46, 208–240. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B 2003, 270, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.; Liu, J.; Sun, C.; Vogler, A.P.; Cai, W. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian site-heterogeneous mixture models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef]
- Li, H.; Leavengood, J.M.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.; Zhou, X.; Cai, W. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. R. Soc. B 2017, 284, 20171223. [Google Scholar] [CrossRef]
- Du, Z.; Hasegawa, H.; Cooley, J.R.; Simon, C.; Yoshimura, J.; Cai, W.; Sota, T.; Li, H. Mitochondrial genomics reveals shared phylogeographic patterns and demographic history among three periodical cicada species groups. Mol. Biol. Evol. 2019, 36, 1187–1200. [Google Scholar] [CrossRef]
- Zhang, D.X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef]
- Clary, D.O.; Wolstenholme, D.R. The mitochondrial DNA molecular of Drosophila yakuba: Nucleotide sequence, gene organization, and genetic code. J. Mol. Evol. 1985, 22, 252–271. [Google Scholar] [CrossRef]
- Shao, R.; Campbell, N.J.H.; Barker, S.C. Numerous gene rearrangements in the mitochondrial genome of the wallaby louse, Heterodoxus macropus (Phthiraptera). Mol. Biol. Evol. 2001, 18, 858–865. [Google Scholar] [CrossRef] [Green Version]
- Shao, R.; Campbell, N.J.H.; Schmidt, E.R.; Barker, S.C. Increased rate of gene rearrangement in the mitochondrial genomes of three orders of hemipteroid insects. Mol. Biol. Evol. 2001, 18, 1828–1832. [Google Scholar] [CrossRef] [Green Version]
- Covacin, C.; Shao, R.; Cameron, S.; Barker, S.C. Extraordinary number of gene rearrangements in the mitochondrial genomes of lice (Phthiraptera: Insecta). Insect Mol. Biol. 2006, 15, 63–68. [Google Scholar] [CrossRef]
- Cameron, S.L.; Johnson, K.P.; Whiting, M.F. The mitochondrial genome of the screamer louse Bothriometopus (Phthiraptera: Ischnocera): Effects of extensive gene rearrangements on the evolution of the genome. J. Mol. Evol. 2007, 65, 589–604. [Google Scholar] [CrossRef]
- Nie, Y.; Fu, Y.T.; Zhang, Y.; Deng, Y.P.; Wang, W.; Tu, Y.; Liu, G.H. Highly rearranged mitochondrial genome in Falcolipeurus lice (Phthiraptera: Philopteridae) from endangered eagles. Parasite. Vector. 2021, 14, 269. [Google Scholar] [CrossRef]
- Kim, K.C. Evolutionary Parallelism in Anoplura and Eutherian Mammals; Oxford University Press: Oxford, UK, 1988. [Google Scholar]
- Price, R.D.; Hellenthal, R.A.; Palma, R.L. World Checklist of Chewing Lice with Host Associations and Keys to Families and Genera; Illinois Natural History Survey Special Publication: Champaign, IL, USA, 2003. [Google Scholar]
- Shao, R.; Kirkness, E.F.; Barker, S.C. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 2009, 19, 904–912. [Google Scholar] [CrossRef] [Green Version]
- Shao, R.; Zhu, X.Q.; Barker, S.C.; Herd, K. Evolution of extensively fragmented mitochondrial genomes in the lice of humans. Genome Biol. Evol. 2012, 4, 1088–1101. [Google Scholar] [CrossRef] [Green Version]
- Song, F.; Li, H.; Liu, G.; Wang, W.; James, P.; Colwell, D.D.; Tran, A.; Gong, S.; Cai, W.; Shao, R. Mitochondrial genome fragmentation unites the parasitic lice of eutherian mammals. Syst. Biol. 2019, 68, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Sweet, A.D.; Johnson, K.P.; Cameron, S.L. Mitochondrial genomes of Columbicola feather lice are highly fragmented, indicating repeated evolution of minicircle-type genomes in parasitic lice. PeerJ 2020, 8, e8759. [Google Scholar] [CrossRef] [Green Version]
- Sweet, A.D.; Johnson, K.P.; Cao, Y.; de Moya, R.S.; Skinner, R.K.; Tan, M.; Herrera, S.V.; Cameron, S.L. Structure, gene order, and nucleotide composition of mitochondrial genomes in parasitic lice from Amblycera. Gene 2021, 768, 145312. [Google Scholar] [CrossRef]
- Fu, Y.T.; Dong, Y.; Wang, W.; Nie, Y.; Liu, G.H.; Shao, R. Fragmented mitochondrial genomes evolved in opposite directions between closely related macaque louse Pedicinus obtusus and colobus louse Pedicinus badii. Genomics 2020, 112, 4924–4933. [Google Scholar] [CrossRef]
- Audi, A.H.; Asmau, A.M. Prevalence of Bird Louse, Menacanthus cornutus (Pthiraptera-Amblycera) in four selected Poultry Farms in Kano State, Nigeria. Bajopas 2014, 7, 142–146. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Kumar, R. Seasonal changes in the population of Menacanthus cornutus (Phthiraptera: Amblycera). J. Parasit. Dis. 2014, 38, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.; Madden, T.; Schäffer, A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Schwentner, M.; Richter, S.; Rogers, D.C.; Giribet, G. Tetraconatan phylogeny with special focus on Malacostraca and Branchiopoda: Highlighting the strength of taxon-specific matrices in phylogenomics. Proc. R. Soc. B 2018, 285, 20181524. [Google Scholar] [CrossRef] [Green Version]
- Song, N.; Li, X.; Yin, X.; Li, X.; Yin, J.; Pan, P. The mitochondrial genomes of palaeopteran insects and insights into the early insect relationships. Sci. Rep. 2019, 9, 17765. [Google Scholar] [CrossRef] [Green Version]
- Lartillot, N.; Rodrigue, N.; Stubbs, D.; Richer, J. PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 2013, 62, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Wu, Y.; Yang, C.; Gu, X.; Wilson, J.J.; Li, H.; Cai, W.; Yang, H.; Song, F. Evolution of tRNA gene rearrangement in the mitochondrial genome of ichneumonoid wasps (Hymenoptera: Ichneumonoidea). Int. J. Biol. Macromol. 2020, 164, 540–547. [Google Scholar] [CrossRef]
- Wei, S.J.; Shi, M.; Chen, X.X.; Sharkey, M.J.; van Achterberg, C.; Ye, G.Y.; He, J.H. New views on strand asymmetry in insect mitochondrial genomes. PLoS ONE 2010, 5, e12708. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.P.; Nguyen, N.; Sweet, A.D.; Boyd, B.M.; Warnow, T.; Allen, J.M. Simultaneous radiation of bird and mammal lice following the K-Pg boundary. Biol. Lett. 2018, 14, 20180141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Moya, R.S.; Yoshizawa, K.; Walden, K.K.O.; Sweet, A.D.; Dietrich, C.H.; Johnson, K.P. Phylogenomics of Parasitic and Nonparasitic Lice (Insecta: Psocodea): Combining Sequence Data and Exploring Compositional Bias Solutions in Next Generation Data Sets. Syst. Biol. 2021, 70, 719–738. [Google Scholar] [CrossRef] [PubMed]
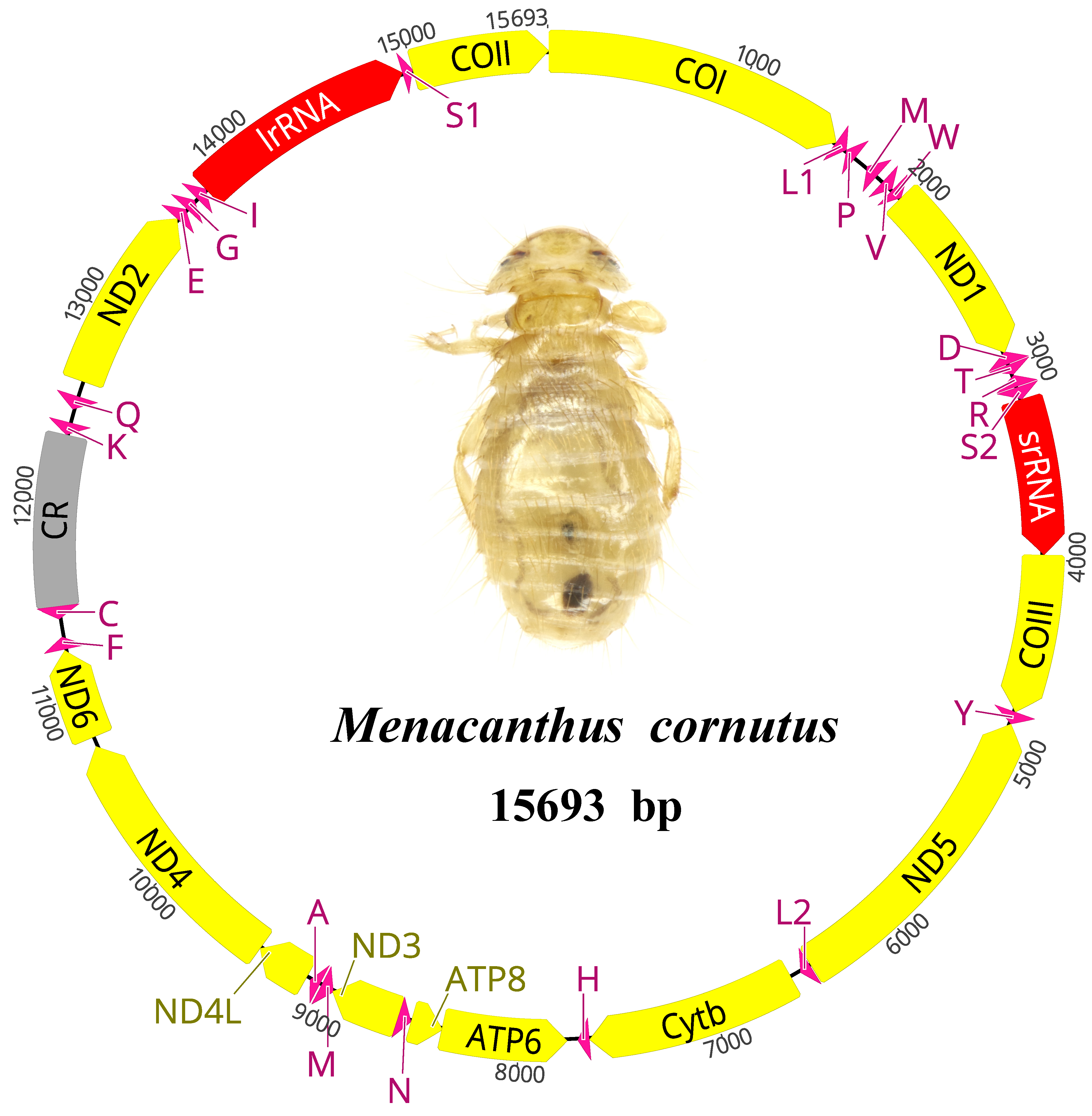



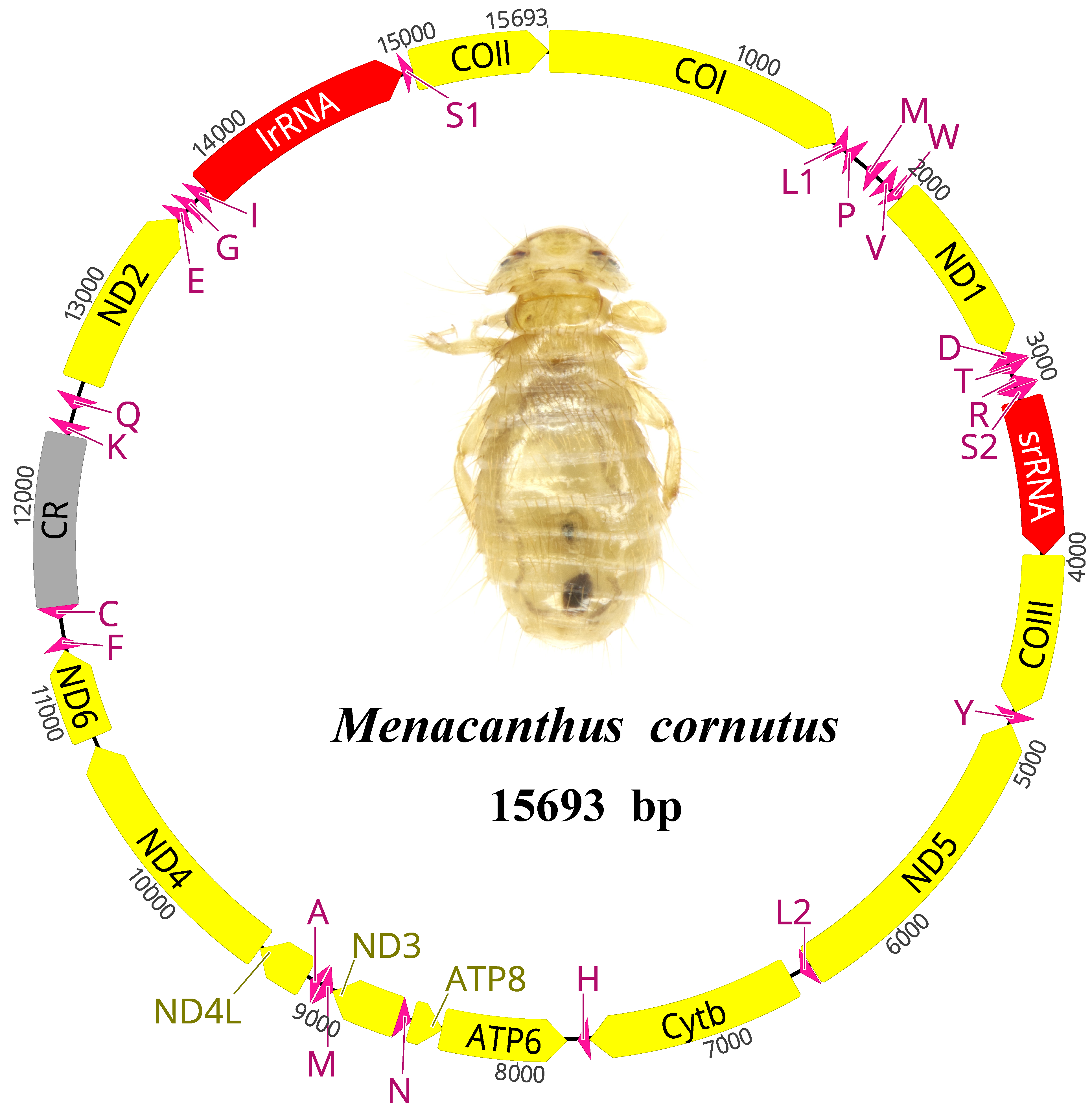{kind=link}
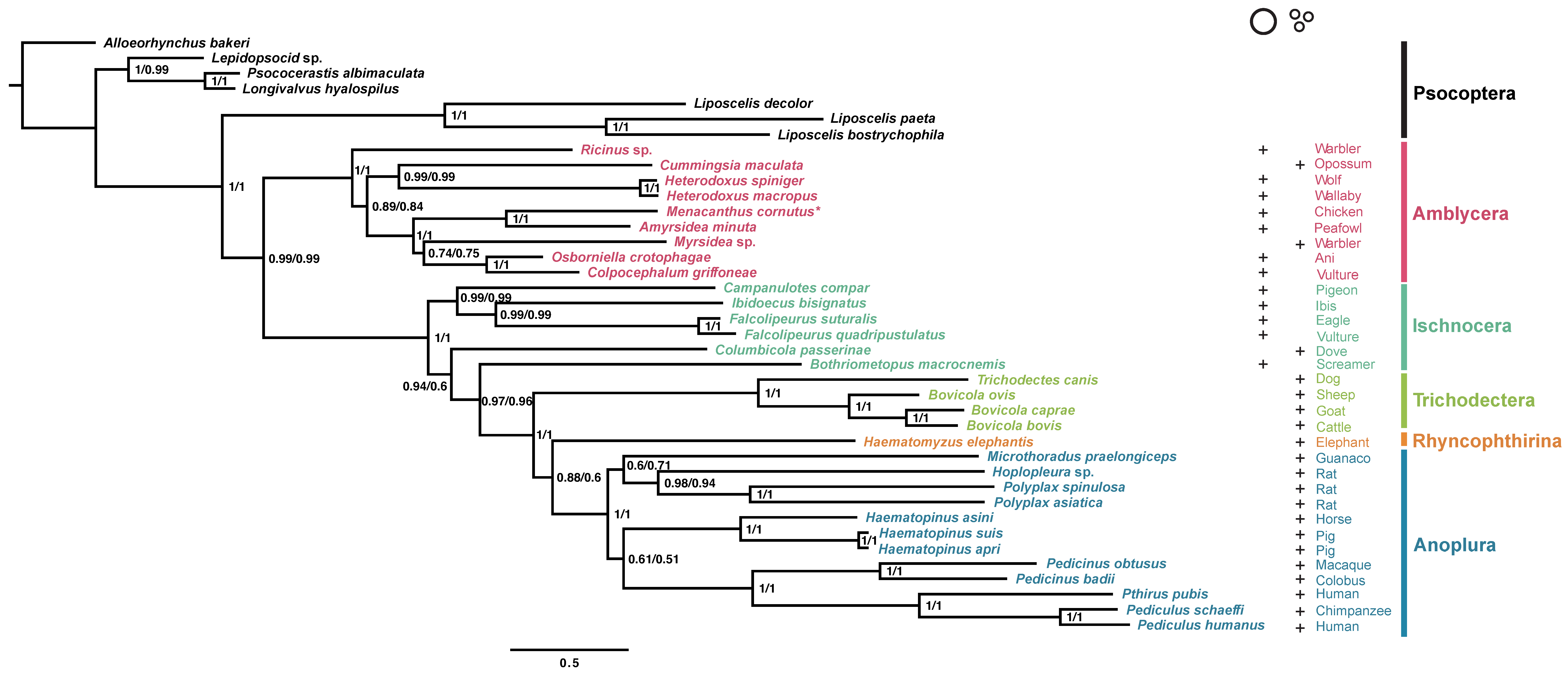{kind=link}
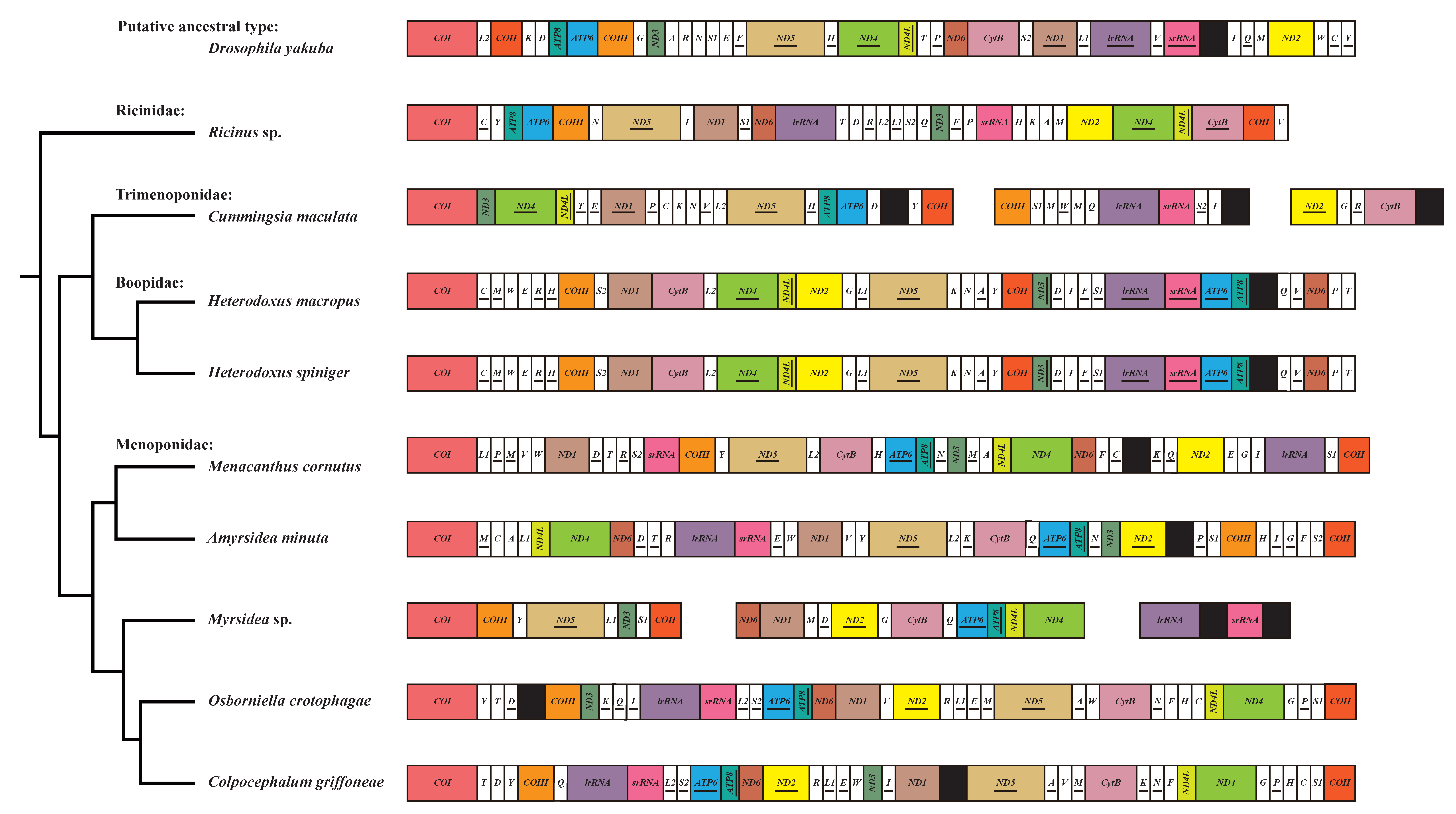{kind=link}
| Order | Suborder | Family | Species | GenBank Accession Number |
|---|---|---|---|---|
| Outgroup | ||||
| Hemiptera | Heteroptera | Nabidae | Alloeorhynchus bakeri | NC_016432 |
| Psocoptera | Trogiomorpha | Lepidopsocidae | Lepidopsocid sp. | NC_004816 |
| Psocomorpha | Psocidae | Psococerastis albimaculata | NC_021400 | |
| Longivalvus hyalospilus | JQ910986 | |||
| Troctomorpha | Liposcelidae | Liposcelis decolor | NC_023839 | |
| Liposcelis bostrychophila | JN645275-645276 | |||
| Liposcelis paeta | KF649225-649226 | |||
| Ingroup | ||||
| Phthiraptera | Amblycera | Boopidae | Heterodoxus macropus | AF270939 |
| Heterodoxus spiniger | MW199168 | |||
| Menoponidae | Menacanthus cornutusa | OM718871 | ||
| Colpocephalum griffoneae | NC_039530 | |||
| Amyrsidea minuta | MH001227 | |||
| Osborniella crotophagae | MW199175 | |||
| Myrsidea sp. | MW199172-199174 | |||
| Ricinidae | Ricinus sp. | MW199176 | ||
| Trimenoponidae | Cummingsia maculata | MW199177-199179 | ||
| Ischnocera | Trichodectidae | Bovicola bovis | MH001189-001200 | |
| Bovicola ovis | MH001201-001212 | |||
| Bovicola caprae | MH001176-001188 | |||
| Trichodectes canis | MH001213-001224 and MH823541 | |||
| Philopteridae | Ibidoecus bisignatus | NC_015999 | ||
| Bothriometopus macrocnemis | EU183542 | |||
| Campanulotes compar | MH001225 | |||
| Falcolipeurus quadripustulatus | MH001226 | |||
| Falcolipeurus suturalis | MW696813 | |||
| Columbicola passerinae | MT094266-094282 | |||
| Rhyncophthirina | Haematomyzidae | Haematomyzus elephantis | KF933032–933041 | |
| Anoplura | Polyplacidae | Polyplax spinulosa | KF647762–647772 | |
| Polyplax asiatica | KF647751–647761 | |||
| Hoplopleuridae | Hoplopleura sp. | MT792483-792494 | ||
| Microthoraciidae | Microthoradus praelongiceps | KX090378–090389 | ||
| Haematopinidae | Haematopinus asini | KF939318, KF939322, KF939324, KF939326, and KJ434034–38 | ||
| Haematopinus apri | KC814611–814619 | |||
| Haematopinus suis | KC814602–814610 | |||
| Pthiridae | Pthirus pubis | EU219987–219995, HM241895–241898, JQ976018, and MT721740 | ||
| Pediculidae | Pediculus schaeffi | KC241882–241897 | ||
| Pediculus humanus | FJ499473–499490 | |||
| Pedicinus obtusus | MT792495–792506 | |||
| Pedicinus badii | MT721726–721739 |
| Family | Species | Fragmented mt Genome | ND4-ND4L | ND4L-ND4 | ATP8-ATP6 | ATP6-ATP8 | trnS1-COII | lrRNA-srRNA |
|---|---|---|---|---|---|---|---|---|
| Drosophila yakuba (Putative ancestral type) | − | + | − | + | − | − | − | |
| Ricinidae | Ricinus sp. | − | + | − | + | − | − | − |
| Trimenoponidae | Cummingsia maculata | + | + | − | + | − | − | + |
| Boopidae | Heterodoxus macropus | − | + | − | − | + | − | − |
| Heterodoxus spiniger | − | + | − | − | + | − | − | |
| Menoponidae | Menacanthus cornutus | − | − | + | − | + | + | − |
| Amyrsidea minuta | − | − | + | − | + | − | + | |
| Myrsidea sp. | + | − | + | − | + | + | + | |
| Osborniella crotophagae | − | − | + | − | + | + | + | |
| Colpocephalum griffoneae | − | − | + | − | + | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, S.; Xu, Y.; Xu, S.; Liang, Y.; Tian, L.; Cai, W.; Li, H.; Song, F. The Complete Mitochondrial Genome of the Chicken Body Louse, Menacanthus cornutus, and Evolutionary Patterns of Extensive Gene Rearrangements in the Mitochondrial Genomes of Amblycera (Psocodea: Phthiraptera). Genes 2022, 13, 522. https://doi.org/10.3390/genes13030522
Gong S, Xu Y, Xu S, Liang Y, Tian L, Cai W, Li H, Song F. The Complete Mitochondrial Genome of the Chicken Body Louse, Menacanthus cornutus, and Evolutionary Patterns of Extensive Gene Rearrangements in the Mitochondrial Genomes of Amblycera (Psocodea: Phthiraptera). Genes. 2022; 13(3):522. https://doi.org/10.3390/genes13030522
Chicago/Turabian StyleGong, Siyu, Ye Xu, Shiwen Xu, Yanxin Liang, Li Tian, Wanzhi Cai, Hu Li, and Fan Song. 2022. "The Complete Mitochondrial Genome of the Chicken Body Louse, Menacanthus cornutus, and Evolutionary Patterns of Extensive Gene Rearrangements in the Mitochondrial Genomes of Amblycera (Psocodea: Phthiraptera)" Genes 13, no. 3: 522. https://doi.org/10.3390/genes13030522
APA StyleGong, S., Xu, Y., Xu, S., Liang, Y., Tian, L., Cai, W., Li, H., & Song, F. (2022). The Complete Mitochondrial Genome of the Chicken Body Louse, Menacanthus cornutus, and Evolutionary Patterns of Extensive Gene Rearrangements in the Mitochondrial Genomes of Amblycera (Psocodea: Phthiraptera). Genes, 13(3), 522. https://doi.org/10.3390/genes13030522