
Identification of Alternative Splicing Events Associated with Paratuberculosis in Dairy Cattle Using Multi-Tissue RNA Sequencing Data




Abstract
:1. Background
2. Materials and Methods
2.1. RNA Sequencing Data
2.2. Quality Control of Reads and Reads Alignment to the Bovine Reference Genome
2.3. Identification of Differentially Expressed Genes
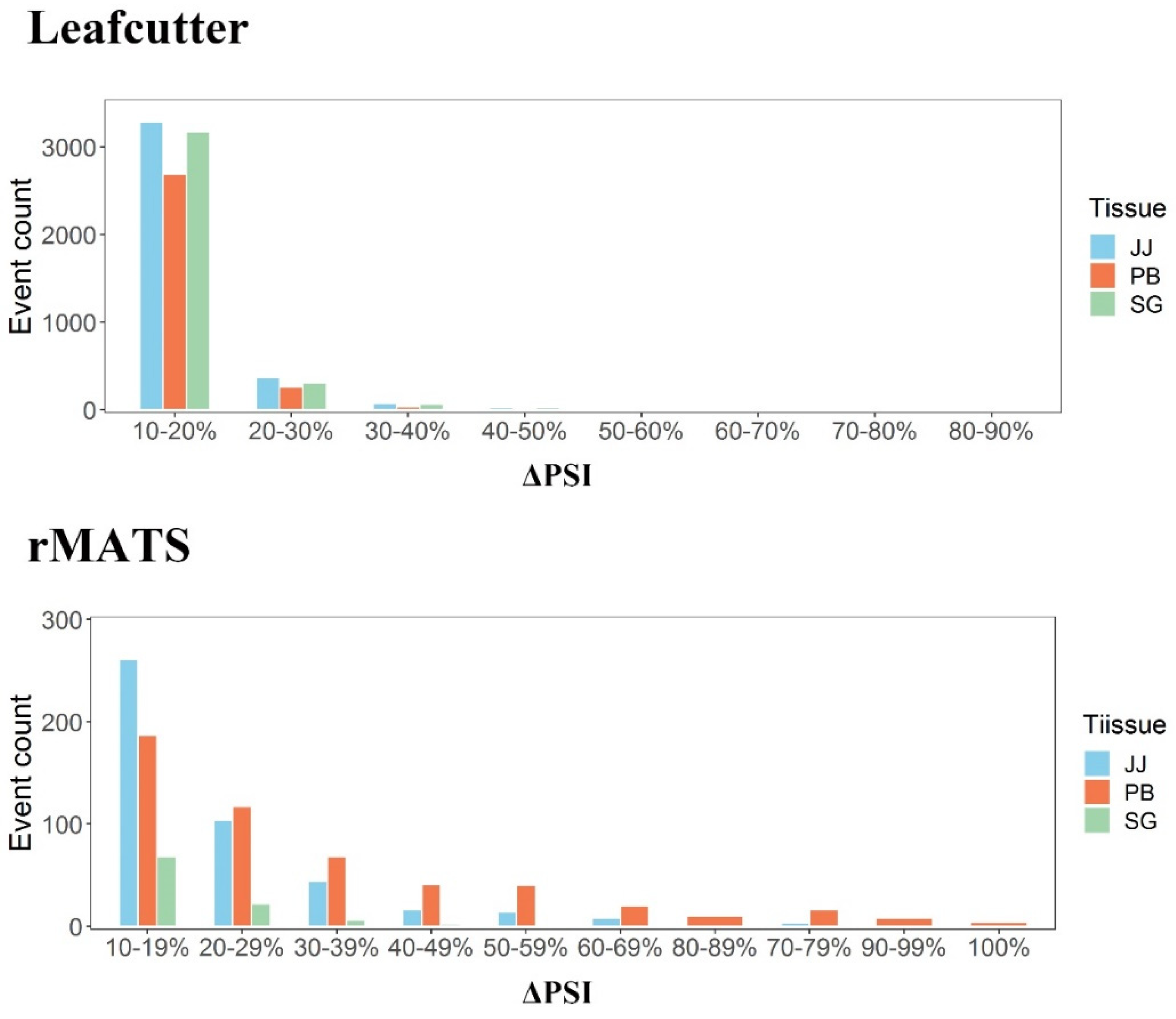
2.4. Identification and Screening of Differential Alternative Splicing (DAS) Events
2.5. Correlation Analysis between Splicing Factors and AS Events
2.6. Statistical Analysis and Protein Structure Prediction
3. Results
3.1. Data Summary
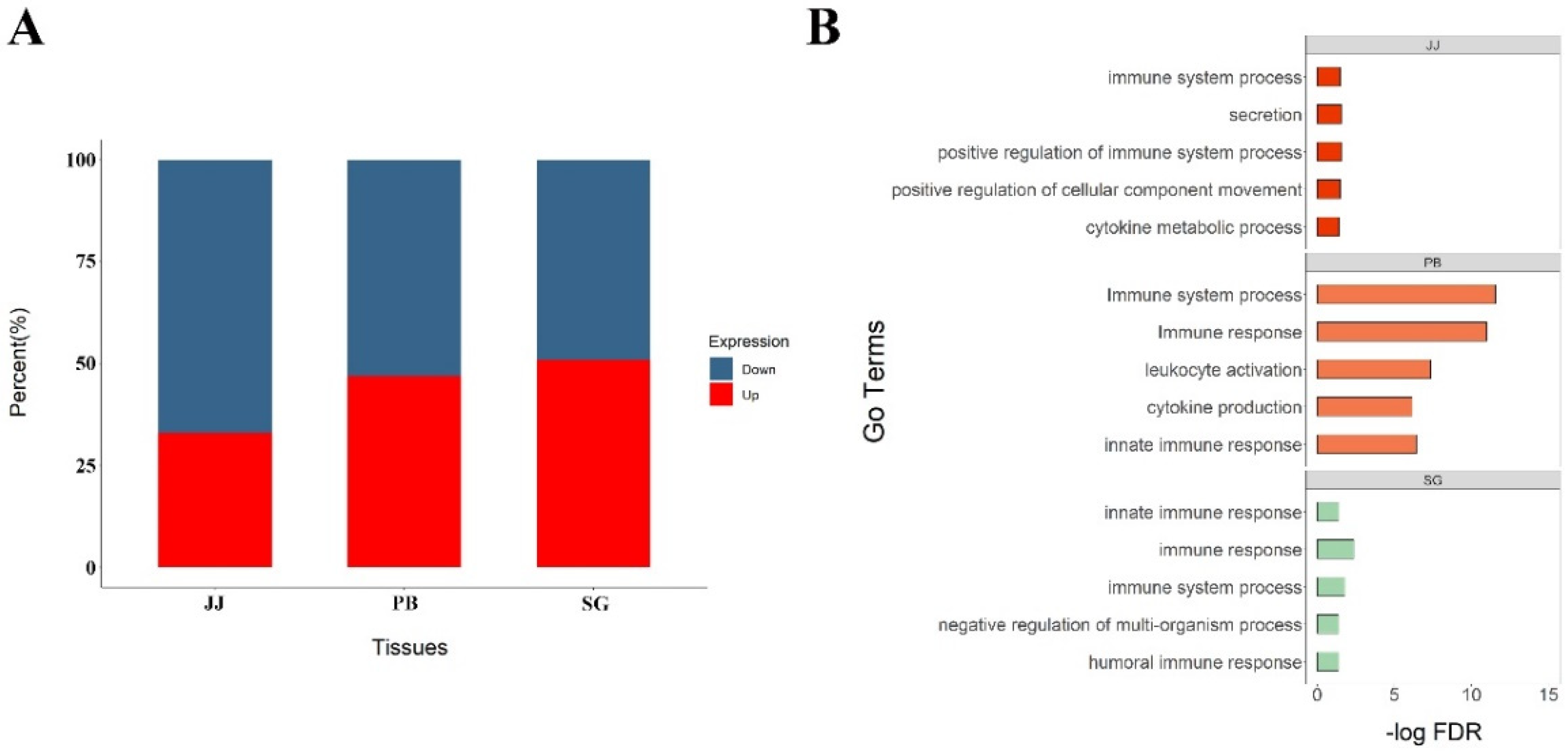
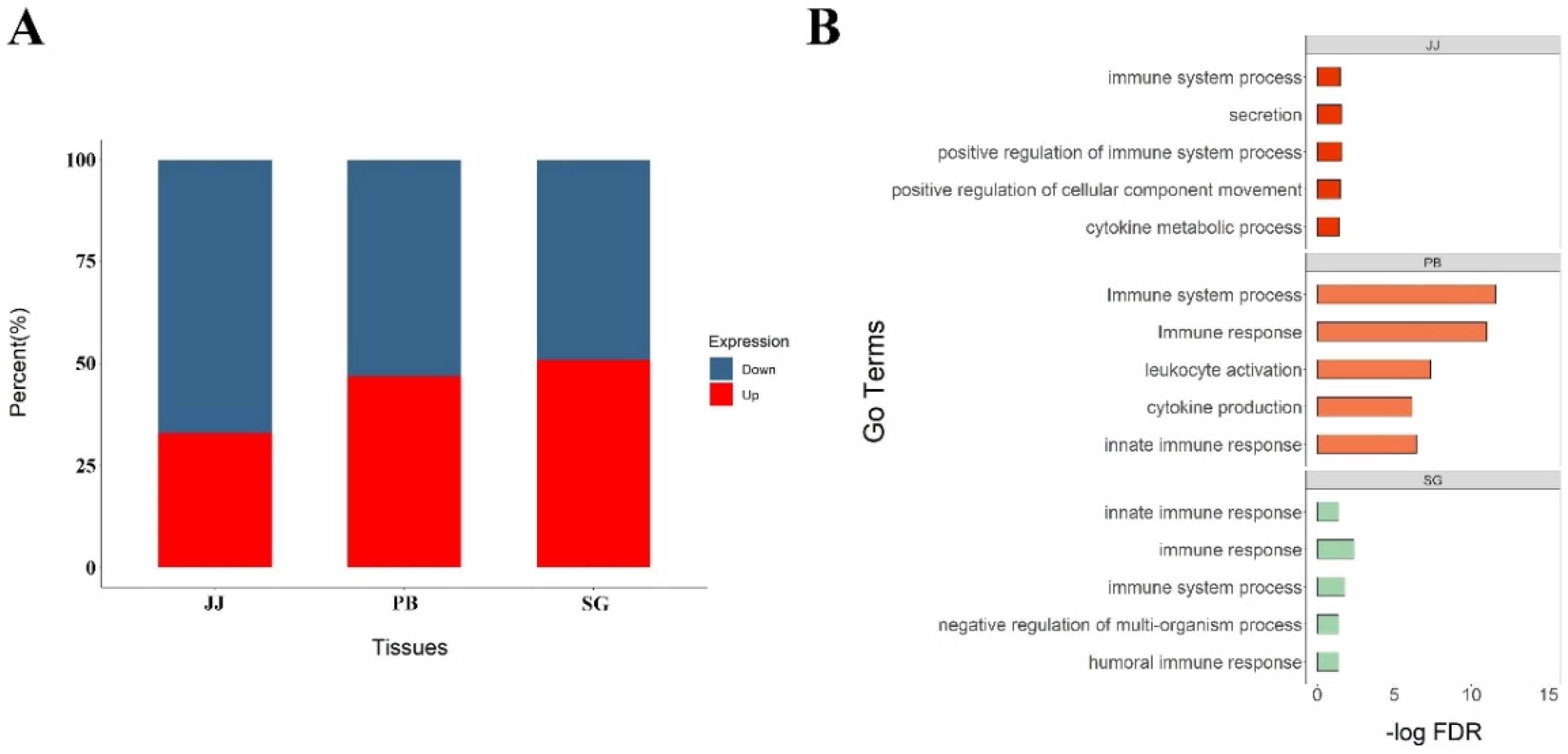
3.2. Differentially Expressed Genes across Tissues
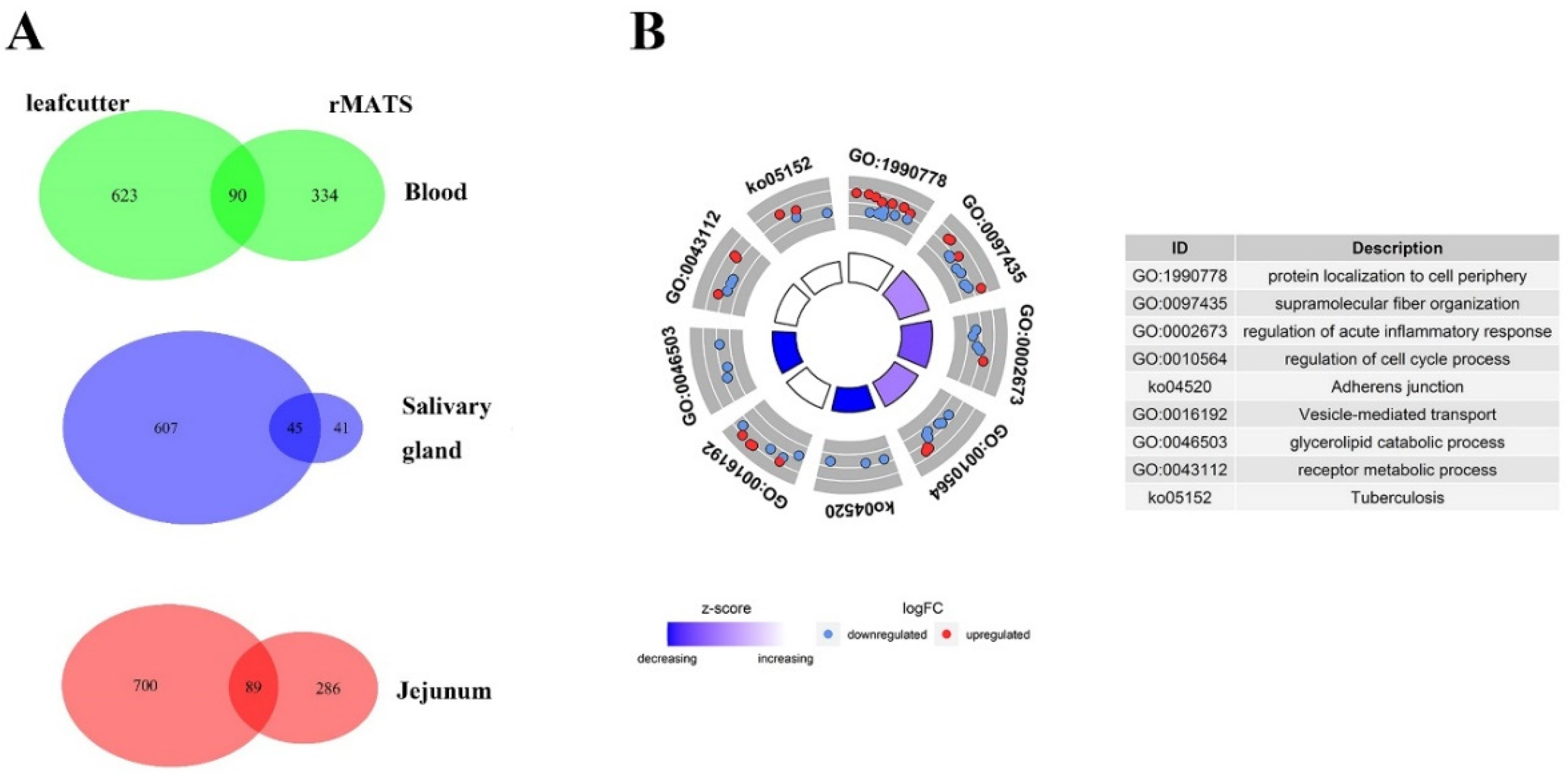
3.3. Differential AS Events
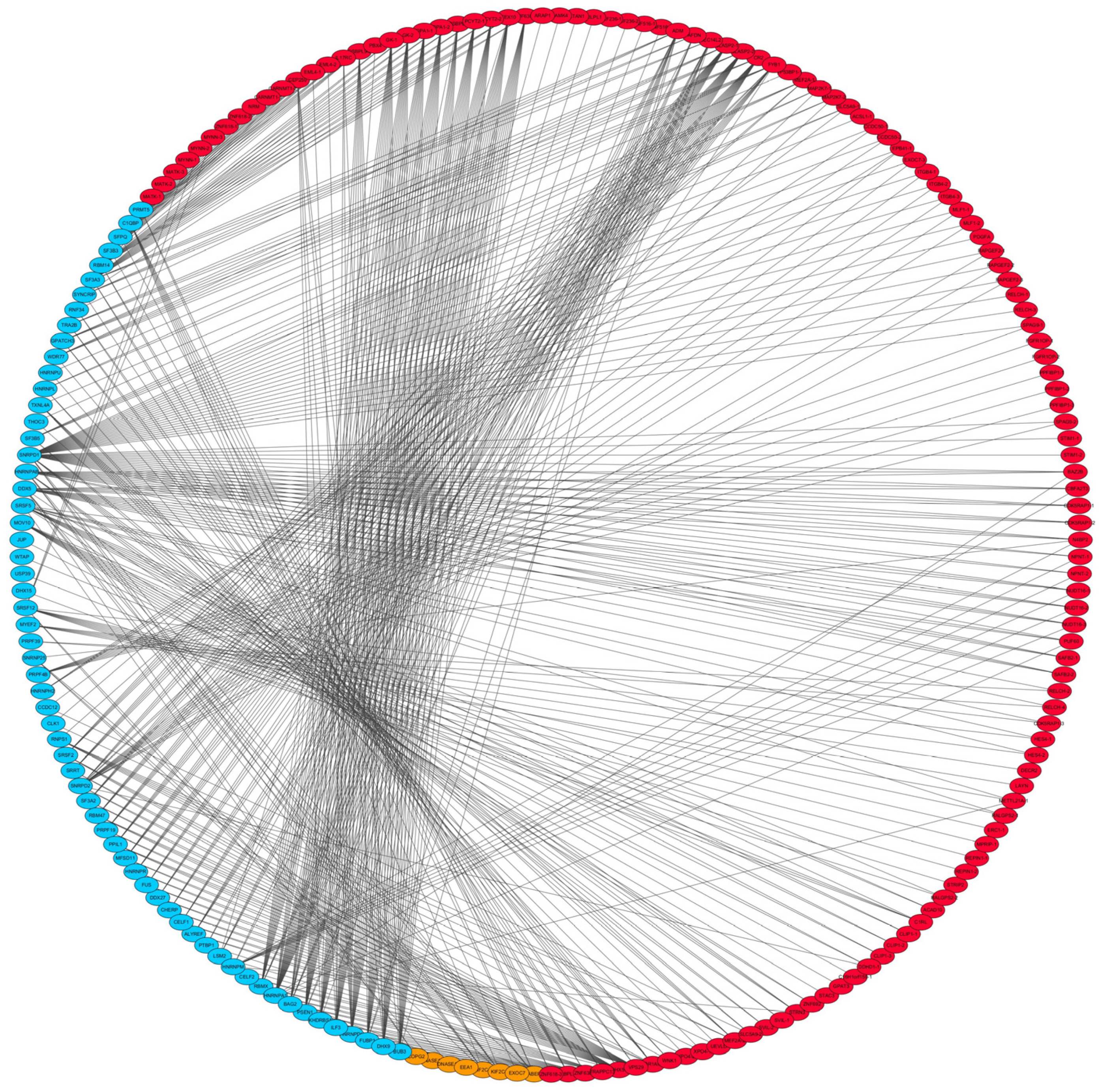
3.4. Correlations between Splicing Factors and DAS Events
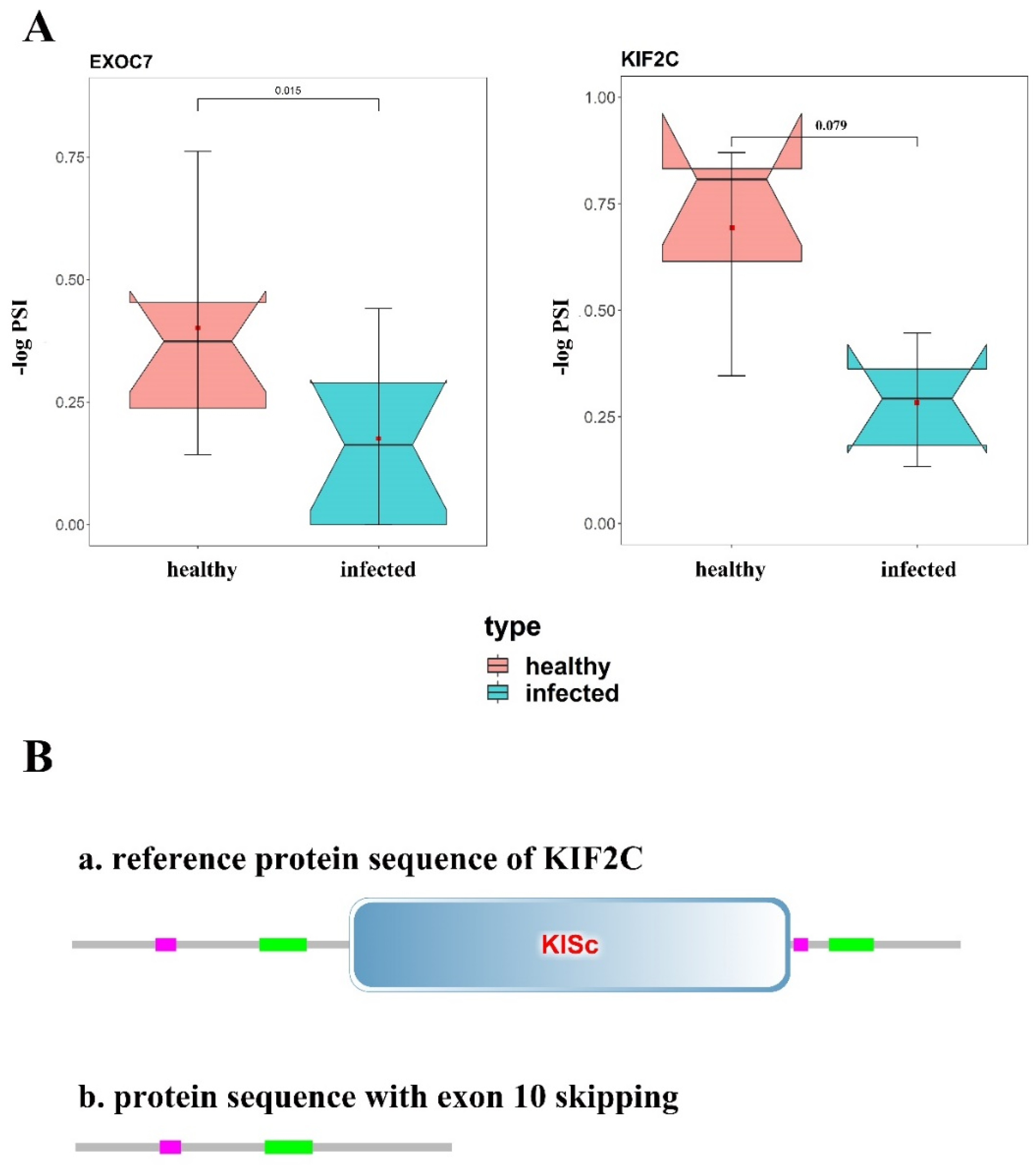
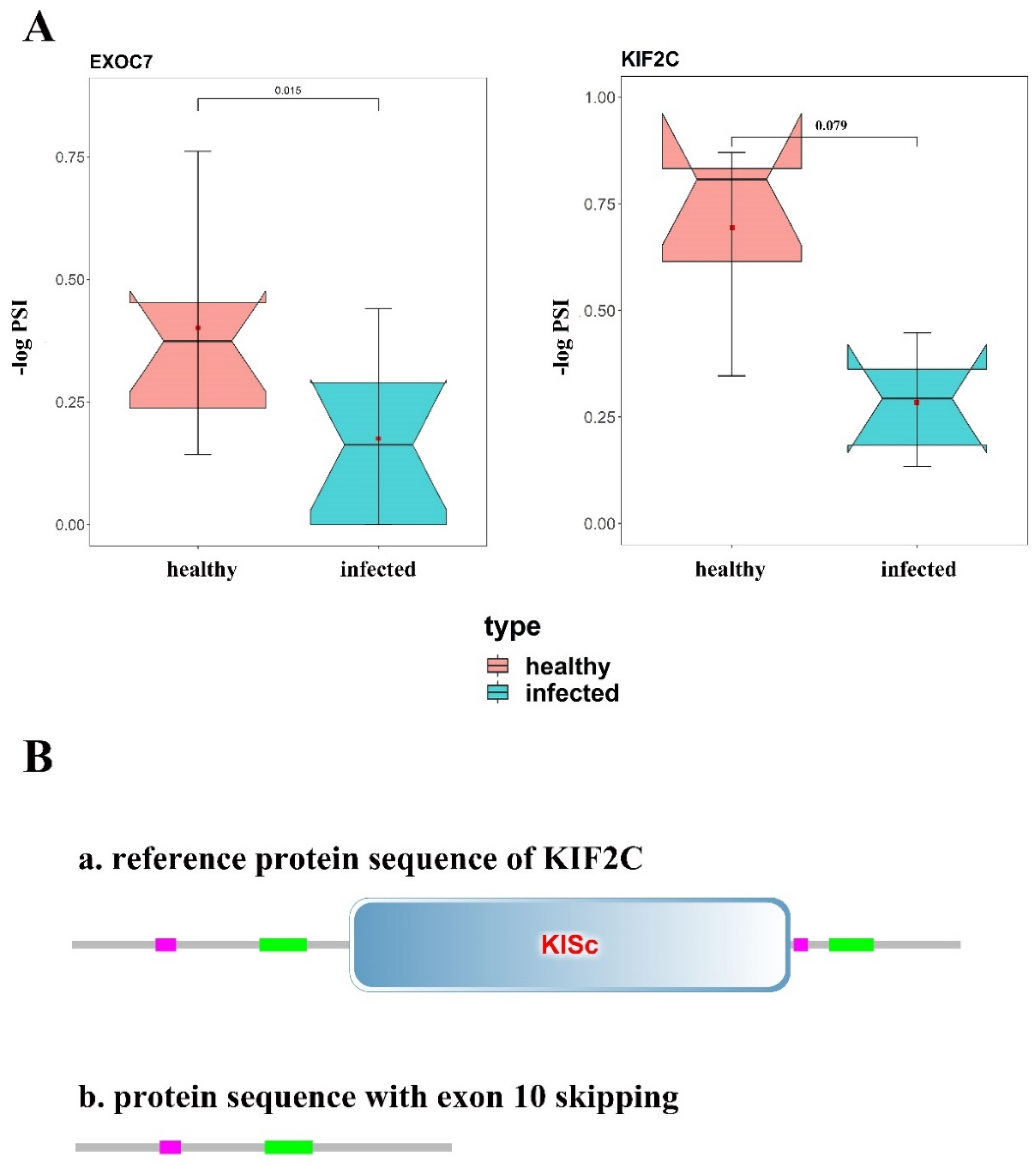
3.5. Changes of Protein Structure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clarke, C.J. The pathology and pathogenesis of paratuberculosis in ruminants and other species. J. Comp. Pathol. 1997, 116, 217–261. [Google Scholar] [CrossRef]
- Stevenson, K.; Alvarez, J.; Bakker, D.; Biet, F.; de Juan, L.; Denham, S.; Dimareli, Z.; Dohmann, K.; Gerlach, G.F.; Heron, I.; et al. Occurrence of Mycobacterium avium subspecies paratuberculosis across host species and European countries with evidence for transmission between wildlife and domestic ruminants. BMC Microbiol. 2009, 9, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, A.B.; Shalloo, L. Invited review: The economic impact and control of paratuberculosis in cattle. J. Dairy Sci. 2015, 98, 5019–5039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.L.; Strawderman, R.L.; Schukken, Y.H.; Wells, S.J.; Pradhan, A.K.; Espejo, L.A.; Whitlock, R.H.; Van Kessel, J.S.; Smith, J.M.; Wolfgang, D.R.; et al. Effect of Johne’s disease status on reproduction and culling in dairy cattle. J. Dairy Sci. 2010, 93, 3513–3524. [Google Scholar] [CrossRef] [PubMed]
- Gonda, M.G.; Kirkpatrick, B.W.; Shook, G.E.; Collins, M.T. Identification of a QTL on BTA20 affecting susceptibility to Mycobacterium avium ssp. paratuberculosis infection in US Holsteins. Anim. Genet. 2007, 38, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.D.; Schenkel, F.S.; Verschoor, C.P.; You, Q.; Kelton, D.F.; Moore, S.S.; Karrow, N.A. A principal component regression based genome wide analysis approach reveals the presence of a novel QTL on BTA7 for MAP resistance in holstein cattle. Genomics 2010, 95, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Kiser, J.N.; White, S.N.; Johnson, K.A.; Hoff, J.L.; Taylor, J.F.; Neibergs, H.L. Identification of loci associated with susceptibility to subspecies tissue infection in cattle. J. Anim. Sci. 2017, 95, 1080–1091. [Google Scholar] [CrossRef]
- Gao, Y.; Jiang, J.; Yang, S.; Cao, J.; Han, B.; Wang, Y.; Zhang, Y.; Yu, Y.; Zhang, S.; Zhang, Q.; et al. Genome-wide association study of Mycobacterium avium subspecies Paratuberculosis infection in Chinese Holstein. BMC Genom. 2018, 19, 972. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y. Identification of Susceptibility/Resistance and Milk Component Trait Functional Genes of Para-Tuberculosis in Dairy Cows Based on Omics Technique; China Agricultural University: Beijing, China, 2017. [Google Scholar]
- Wilkinson, M.E.; Charenton, C.; Nagai, K. RNA Splicing by the Spliceosome. Annu. Rev. Biochem. 2020, 89, 359–388. [Google Scholar] [CrossRef]
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Li, K.; Zhao, W.; Cui, Q. SpliceDisease database: Linking RNA splicing and disease. Nucleic Acids Res. 2012, 40, D1055–D1059. [Google Scholar] [CrossRef] [PubMed]
- Montes, M.; Sanford, B.L.; Comiskey, D.F.; Chandler, D.S. RNA Splicing and Disease: Animal Models to Therapies. Trends Genet. TIG 2019, 35, 68–87. [Google Scholar] [CrossRef] [PubMed]
- Wobst, H.J.; Denk, F.; Oliver, P.L.; Livieratos, A.; Taylor, T.N.; Knudsen, M.H.; Bengoa-Vergniory, N.; Bannerman, D.; Wade-Martins, R. Increased 4R tau expression and behavioural changes in a novel MAPT-N296H genomic mouse model of tauopathy. Sci. Rep. 2017, 7, 43198. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999. [Google Scholar] [CrossRef]
- Liu, S.M.; Sutherland, A.P.; Zhang, Z.; Rainbow, D.B.; Quintana, F.J.; Paterson, A.M.; Sharpe, A.H.; Oukka, M.; Wicker, L.S.; Kuchroo, V.K. Overexpression of the Ctla-4 isoform lacking exons 2 and 3 causes autoimmunity. J. Immunol. 2012, 188, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.G.; Ju, Z.H.; Hou, M.H.; Jiang, Q.; Yang, C.H.; Zhang, Y.; Sun, Y.; Li, R.L.; Wang, C.F.; Zhong, J.F.; et al. Deciphering Transcriptome and Complex Alternative Splicing Transcripts in Mammary Gland Tissues from Cows Naturally Infected with Staphylococcus aureus Mastitis. PLoS ONE 2016, 11, e0159719. [Google Scholar] [CrossRef]
- Yang, L.; Guo, R.; Ju, Z.; Wang, X.; Jiang, Q.; Liu, Y.; Zhao, H.; He, K.; Li, J.; Huang, J. Production of an aberrant splice variant of CCL5 is not caused by genetic mutation in the mammary glands of mastitis-infected Holstein cows. Mol. Med. Rep. 2019, 19, 4159–4166. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Malmuthuge, N.; Guan, Y.; Ren, Y.; Griebel, P.J.; le Guan, L. Altered microRNA expression and pre-mRNA splicing events reveal new mechanisms associated with early stage Mycobacterium avium subspecies paratuberculosis infection. Sci. Rep. 2016, 6, 24964. [Google Scholar] [CrossRef] [Green Version]
- Emmert, D.B.; Stoehr, P.J.; Stoesser, G.; Cameron, G.N. The European Bioinformatics Institute (EBI) databases. Nucleic Acids Res. 1994, 22, 3445–3449. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Cao, J.; Zhang, S.; Zhang, Q.; Sun, D. Short communication: Heritability estimates for susceptibility to Mycobacterium avium ssp. paratuberculosis infection in Chinese Holstein cattle. J. Dairy Sci. 2018, 101, 7274–7279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso-Hearn, M.; Canive, M.; Blanco-Vazquez, C.; Torremocha, R.; Balseiro, A.; Amado, J.; Varela-Martinez, E.; Ramos, R.; Jugo, B.M.; Casais, R. RNA-Seq analysis of ileocecal valve and peripheral blood from Holstein cattle infected with Mycobacterium avium subsp. paratuberculosis revealed dysregulation of the CXCL8/IL8 signaling pathway. Sci. Rep. 2019, 9, 14845. [Google Scholar] [CrossRef] [PubMed]
- Mallikarjunappa, S.; Adnane, M.; Cormican, P.; Karrow, N.A.; Meade, K.G. Characterization of the bovine salivary gland transcriptome associated with Mycobacterium avium subsp. paratuberculosis experimental challenge. BMC Genom. 2019, 20, 491. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Schlapbach, R.; Rehrauer, H. RNA-Seq Data Analysis from Raw Data Quality Control to Differential Expression Analysis. Methods Mol. Biol. 2017, 1669, 295–307. [Google Scholar]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. GigaScience 2020, 9, 3. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.I.; Knowles, D.A.; Humphrey, J.; Barbeira, A.N.; Dickinson, S.P.; Im, H.K.; Pritchard, J.K. Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet. 2018, 50, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.J.; Byun, S.; Han, S.; Chamberlin, J.; Kim, D.; Kim, M.J.; Lee, Y. Differential alternative splicing regulation among hepatocellular carcinoma with different risk factors. BMC Med. Genom. 2019, 12 (Suppl. 8), 175. [Google Scholar] [CrossRef]
- Du, J.X.; Zhu, G.Q.; Cai, J.L.; Wang, B.; Luo, Y.H.; Chen, C.; Cai, C.Z.; Zhang, S.J.; Zhou, J.; Fan, J.; et al. Splicing factors: Insights into their regulatory network in alternative splicing in cancer. Cancer Lett. 2021, 501, 83–104. [Google Scholar] [CrossRef]
- Giulietti, M.; Piva, F.; D’Antonio, M.; D’Onorio De Meo, P.; Paoletti, D.; Castrignanò, T.; D’Erchia, A.M.; Picardi, E.; Zambelli, F.; Principato, G.; et al. SpliceAid-F: A database of human splicing factors and their RNA-binding sites. Nucleic Acids Res. 2013, 41, D125–D131. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [Green Version]
- Massey, F.J., Jr. The Kolmogorov-Smirnov test for goodness of fit. J. Am. Stat. Assoc. 1951, 46, 68–78. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators: Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46(D1), D8–D13.
- Schultz, J.; Copley, R.R.; Doerks, T.; Ponting, C.P.; Bork, P. SMART: A web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000, 28, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Bannantine, J.P.; Bermudez, L.E. No holes barred: Invasion of the intestinal mucosa by Mycobacterium avium subsp. paratuberculosis. Infect. Immun. 2013, 81, 3960–3965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsenault, R.J.; Maattanen, P.; Daigle, J.; Potter, A.; Griebel, P.; Napper, S. From mouth to macrophage: Mechanisms of innate immune subversion by Mycobacterium avium subsp. paratuberculosis. Vet. Res. 2014, 45, 54. [Google Scholar] [CrossRef] [Green Version]
- Määttänen, P.; Trost, B.; Scruten, E.; Potter, A.; Kusalik, A.; Griebel, P.; Napper, S. Divergent immune responses to Mycobacterium avium subsp. paratuberculosis infection correlate with kinome responses at the site of intestinal infection. Infect. Immun. 2013, 81, 2861–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, J.M.; Rankin, J.D. The Pathogenesis of Experimental Johne’s Disease in Calves. Res. Vet. Sci. 1961, 2, 167–176. [Google Scholar] [CrossRef]
- Sweeney, R.W.; Uzonna, J.; Whitlock, R.H.; Habecker, P.L.; Chilton, P.; Scott, P. Tissue predilection sites and effect of dose on Mycobacterium avium subs. paratuberculosis organism recovery in a short-term bovine experimental oral infection model. Res. Vet. Sci. 2006, 80, 253–259. [Google Scholar] [CrossRef]
- Fábián, T.K.; Hermann, P.; Beck, A.; Fejérdy, P.; Fábián, G. Salivary defense proteins: Their network and role in innate and acquired oral immunity. Int. J. Mol. Sci. 2012, 13, 4295–4320. [Google Scholar] [CrossRef]
- Mathews, M.; Jia, H.P.; Guthmiller, J.M.; Losh, G.; Graham, S.; Johnson, G.K.; Tack, B.F.; McCray, P.B., Jr. Production of β-defensin antimicrobial peptides by the oral mucosa and salivary glands. Infect. Immun. 1999, 67, 2740–2745. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Delcher, A.L.; Florea, L.; Kelley, D.R.; Schatz, M.C.; Puiu, D.; Hanrahan, F.; Pertea, G.; Van Tassell, C.P.; Sonstegard, T.S.; et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009, 10, R42. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Crisman, L.; Miller, J.; Datta, I.; Gulbranson, D.R.; Tian, Y.; Yin, Q.; Yu, H.; Shen, J. Inducible Exoc7/Exo70 knockout reveals a critical role of the exocyst in insulin-regulated GLUT4 exocytosis. J. Biol. Chem. 2019, 294, 19988–19996. [Google Scholar] [CrossRef]
- Georgilis, A.; Klotz, S.; Hanley, C.J.; Herranz, N.; Weirich, B.; Morancho, B.; Leote, A.C.; D’Artista, L.; Gallage, S.; Seehawer, M.; et al. PTBP1-Mediated Alternative Splicing Regulates the Inflammatory Secretome and the Pro-tumorigenic Effects of Senescent Cells. Cancer Cell 2018, 34, 85–102.e109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Liu, J.; Liu, S.; Li, W. Identification of Crucial Genes Associated With Immune Cell Infiltration in Hepatocellular Carcinoma by Weighted Gene Co-expression Network Analysis. Front. Genet. 2020, 11, 342. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Zhang, J.; Feng, D.; Zhao, Y.; Ouyang, W.; Shi, R.; Zhou, X.; Yu, Z.; Wei, S.; Min, J.; et al. KIF2C Is a Novel Prognostic Biomarker and Correlated with Immune Infiltration in Endometrial Cancer. Stem Cells Int. 2021, 2021, 1434856. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, A.; Laiho, A.; Venäläinen, M.S.; McGlinchey, A.J.; Wang, N.; Elo, L.L. Systematic evaluation of differential splicing tools for RNA-seq studies. Brief. Bioinform. 2020, 21, 2052–2065. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait Material | Data Grouping | Sequencing Platform | Breed | Data Sources | Accession Numbers |
|---|---|---|---|---|---|
| Jejunum | NP, n = 3 DP, n = 3 | Illumina Hiseq 2500 (Bos taurus) | Chinese Holstein cows | Gao Yahui, 2017 | PRJNA756737 |
| Peripheral blood | control, n = 2 case, n = 2 | Illumina NovaSeq 6000 (Bos taurus) | Korean Holstein cows | Seoul National University, Park H, Yoo H, 2020 | PRJNA628877 |
| Salivary Gland | control, n = 7 case, n = 7 | Illumina NovaSeq 6000 (Bos taurus) | Irish Holstein cows | University of Guelph, Mallikarjunappa S, 2019 | PRJNA513864 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Huang, J.; Zhang, J.; Gao, Y.; Han, B.; Sun, D. Identification of Alternative Splicing Events Associated with Paratuberculosis in Dairy Cattle Using Multi-Tissue RNA Sequencing Data. Genes 2022, 13, 497. https://doi.org/10.3390/genes13030497
Li H, Huang J, Zhang J, Gao Y, Han B, Sun D. Identification of Alternative Splicing Events Associated with Paratuberculosis in Dairy Cattle Using Multi-Tissue RNA Sequencing Data. Genes. 2022; 13(3):497. https://doi.org/10.3390/genes13030497
Chicago/Turabian StyleLi, Houcheng, Jinfeng Huang, Junnan Zhang, Yahui Gao, Bo Han, and Dongxiao Sun. 2022. "Identification of Alternative Splicing Events Associated with Paratuberculosis in Dairy Cattle Using Multi-Tissue RNA Sequencing Data" Genes 13, no. 3: 497. https://doi.org/10.3390/genes13030497
APA StyleLi, H., Huang, J., Zhang, J., Gao, Y., Han, B., & Sun, D. (2022). Identification of Alternative Splicing Events Associated with Paratuberculosis in Dairy Cattle Using Multi-Tissue RNA Sequencing Data. Genes, 13(3), 497. https://doi.org/10.3390/genes13030497