
Recombination Mediator Proteins: Misnomers That Are Key to Understanding the Genomic Instabilities in Cancer


{kind=link}
{kind=link}
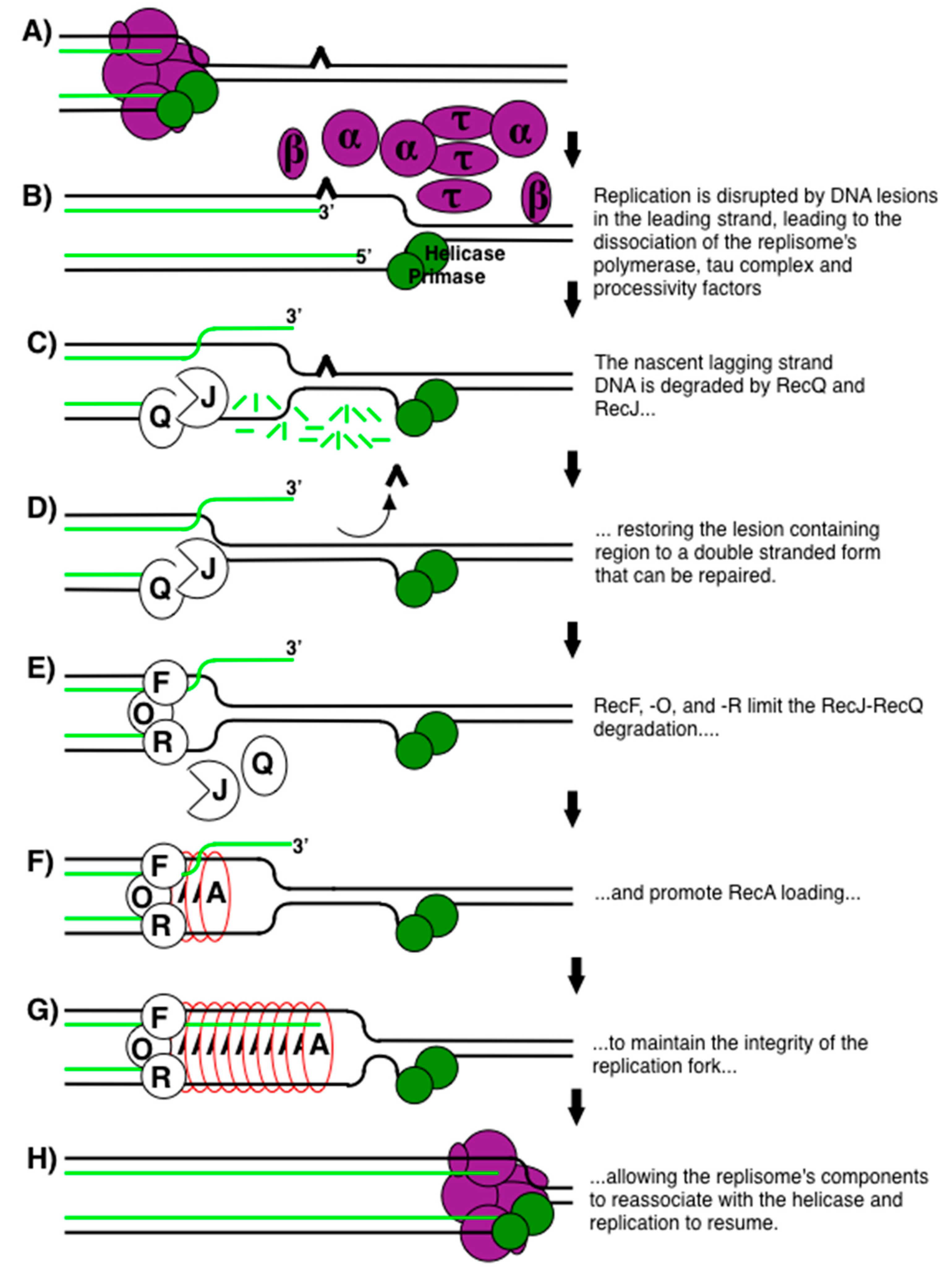{kind=link}
{kind=link}
{kind=link}
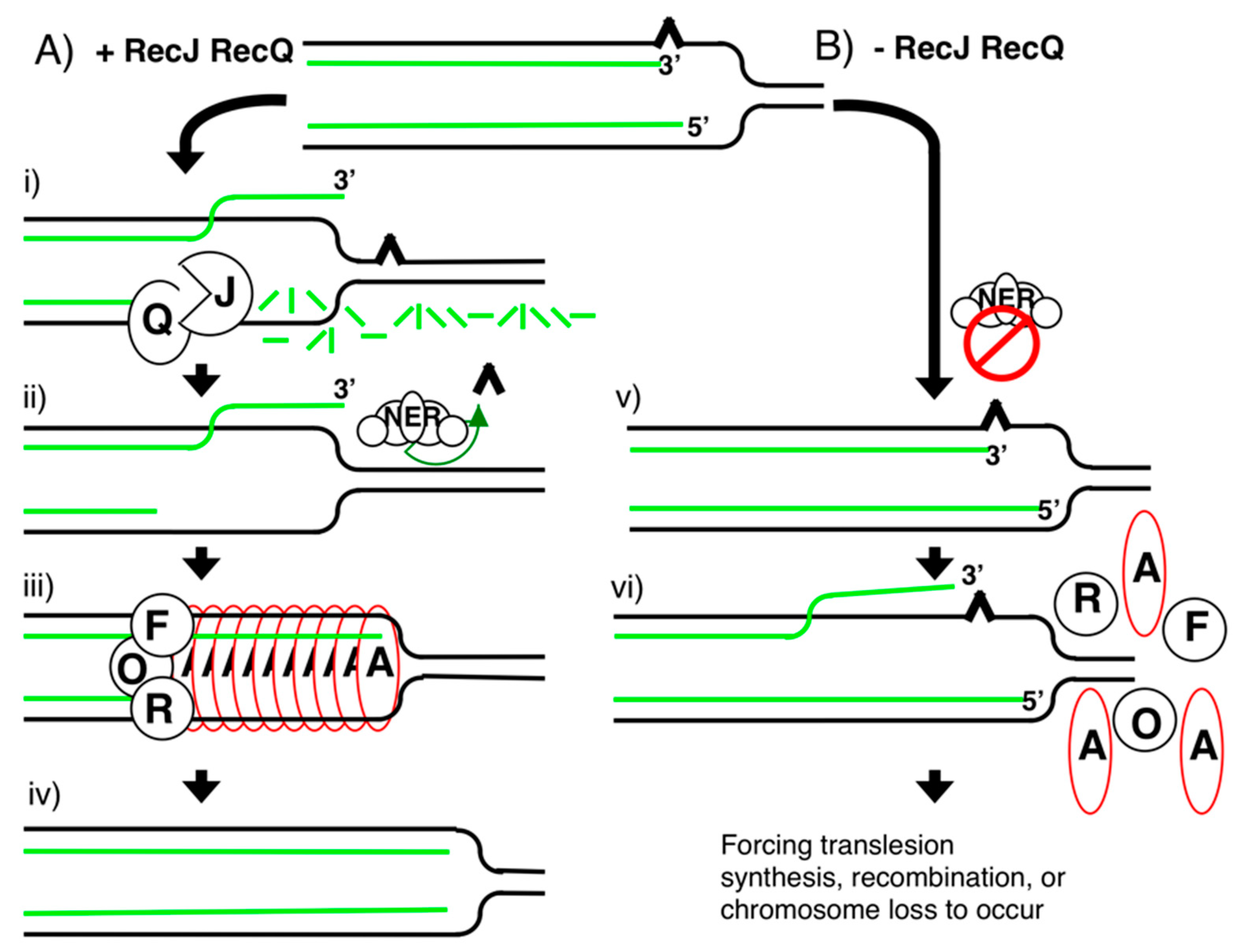{kind=link}
{kind=link}
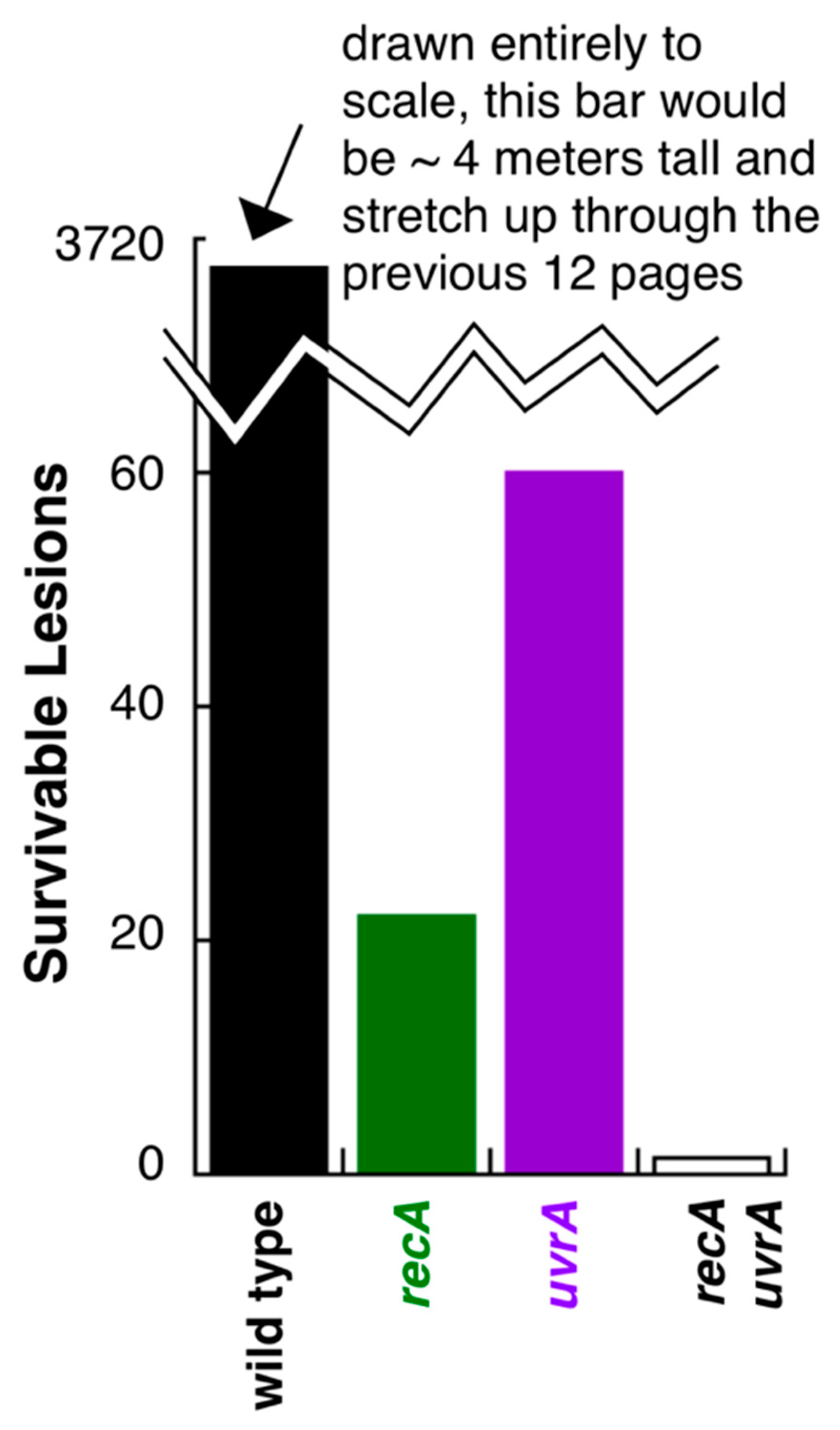{kind=link}
{kind=link}
{kind=link}
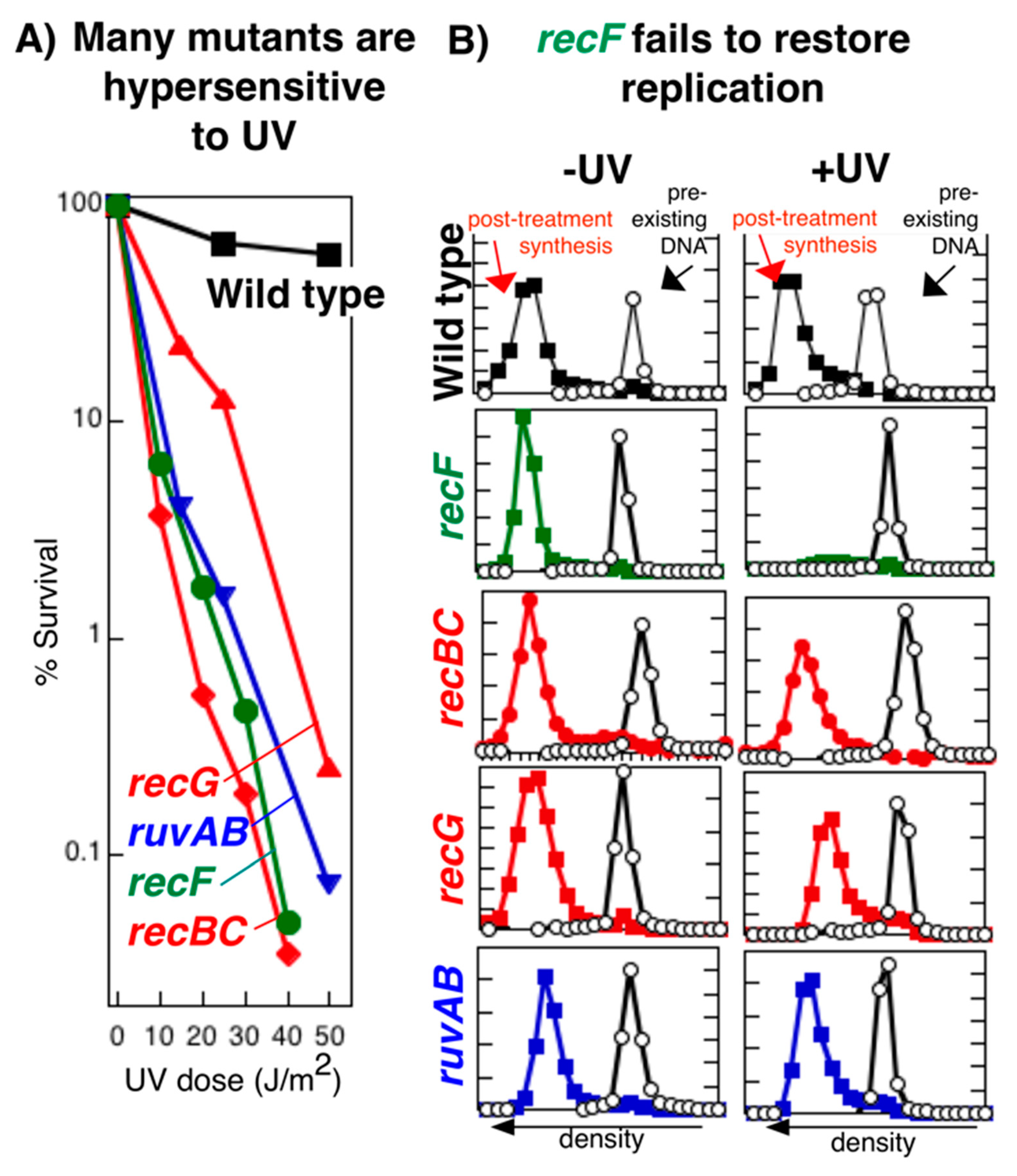{kind=link}
{kind=link}
Abstract
Contents
- Nonrecombinational roles of recombination mediators during replication
- Replication substrates created during encounters with DNA damage
- The RecF pathway maintains replication following disruption by DNA damage
- RecF, RecO, and RecR are required to maintain the replication fork and restore processive replication
- RecJ and RecQ process the replication forks following DNA damage to promote resumption from the site where disruption occurred
- Evidence that lesion repair occurs at disrupted replication forks to prevent mutations or rearrangements by translesion synthesis or recombination
- Maintaining and restoring processive replication is specific to the RecF pathway
- Recombination mediators are functionally conserved between bacteria and mammals
1. Nonrecombinational Roles of Recombination Mediators during Replication
2. Replication Substrates Created during Encounters with DNA Damage
3. The RecF Pathway Maintains Replication Following Disruption by DNA Damage
4. RecF, RecO, and RecR Are Required to Maintain the Replication Fork and Restore Processive Replication
5. RecJ and RecQ Process the Replication Forks Following DNA Damage to Promote Resumption from the Site Where Disruption Occurred
6. Evidence That Lesion Repair Occurs at Disrupted Replication Forks to Prevent Mutations or Rearrangements by Translesion Synthesis or Recombination
7. Maintaining and Restoring Processive Replication Is Specific to the RecF Pathway
8. Recombination Mediators Are Functionally Conserved between Bacteria and Mammals
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lederberg, J.; Tatum, E.L. Sex in Bacteria: Genetic Studies, 1945–1952. Science 1953, 118, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Margulies, A.D. Isolation and characterization of recombination-deficient mutants of Escherichia coli K12. Proc. Natl. Acad. Sci. USA 1965, 53, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J. The beginning of a genetic analysis of recombination proficiency. J. Cell. Physiol. 1967, 70, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Horii, Z.; Clark, A.J. Genetic analysis of the recF pathway to genetic recombination in Escherichia coli K12: Isolation and characterization of mutants. J. Mol. Biol. 1973, 80, 327–344. [Google Scholar] [CrossRef]
- Howard-Flanders, P.; Theriot, L. Mutants of Escherichia coli K-12 defective in DNA repair and in genetic recombination. Genetics 1966, 53, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Rupp, W.D.; Howard-Flanders, P. Discontinuities in the DNA synthesized in an Excision-defective strain of Escherichia coli following ultraviolet irradiation. J. Mol. Biol. 1968, 31, 291–304. [Google Scholar] [CrossRef]
- Rupp, W.D.; Wilde, C.E.; Reno, D.L.; Howard-Flanders, P. Exchanges between DNA strands in ultraviolet-irradiated Escherichia coli. J. Mol. Biol. 1971, 61, 25–44. [Google Scholar] [CrossRef]
- Dhar, P.K.; Devi, S.; Rao, T.R.; Kumari, U.; Joseph, A.; Kumar, M.; Nayak, S.; Shreemati, Y.; Bhat, S.; Bhat, K.M. Significance of lymphocytic sister chromatid exchange frequencies in ovarian cancer patients. Cancer Genet. Cytogenet. 1996, 89, 105–108. [Google Scholar] [CrossRef]
- Dönmez, H.; Özkul, Y.; Uçak, R. Sister chromatid exchange frequency in inhabitants exposed to asbestos in Turkey. Mutat. Res. Mutagen. Relat. Subj. 1996, 361, 129–132. [Google Scholar] [CrossRef]
- Murthy, M.K.; Bhargava, M.K.; Augustus, M. Sister chromatid exchange studies in oral cancer patients. Indian J. Cancer 1997, 34, 49–58. [Google Scholar]
- Husain, S.; Balasubramanian, S.; Bamezai, R. Sister chromatid exchange frequency in breast cancer cases. Cancer Genet. Cytogenet. 1992, 61, 142–146. [Google Scholar] [CrossRef]
- Dhillon, V.S.; Bhasker, R.; Kler, R.S.; Husain, S.A. Sister chromatid exchange (SCE) studies in breast cancer patients: A follow-up study. Cancer Genet. Cytogenet. 1995, 80, 115–117. [Google Scholar] [CrossRef]
- Wang, L.Y.; Lai, M.S.; Huang, S.J.; Hsieh, C.Y.; Hsu, M.M.; Chen, C.J. Increased sister chromatid exchange frequency in peripheral lymphocytes of nasopharyngeal carcinoma and cervical cancer patients. Anticancer Res. 1994, 14, 105–107. [Google Scholar] [PubMed]
- Dhillon, V.S.; Kler, R.S.; Dhillon, I.K. Choromosome instabililty and sister chromatid exchange (SCE) studies in patients with carcinoma of cervix uteri. Cancer Genet. Cytogenet. 1996, 86, 54–57. [Google Scholar] [CrossRef]
- Xu, X.; Wiencke, J.K.; Niu, T.; Wang, M.; Watanabe, H.; Kelsey, K.T.; Christiani, D.C. Benzene exposure, glutathione S-transferase theta homozygous deletion, and sister chromatid exchanges. Am. J. Ind. Med. 1998, 33, 157–163. [Google Scholar] [CrossRef]
- Strom, S.S.; Gu, Y.; Sigurdson, A.J.; Bailey, N.M.; Amos, C.I.; Spitz, M.R.; Rodriguez, M.A.; Liang, J.C. Chromosome breaks and sister chromatid exchange as predictors of second cancers in Hodgkin’s disease. Leuk. Lymphoma 1998, 28, 561–566. [Google Scholar] [CrossRef]
- Hanada, K.; Ukita, T.; Kohno, Y.; Saito, K.; Kato, J.-I.; Ikeda, H. RecQ DNA helicase is a suppressor of illegitimate recombination in Escherichia coli. Proc. Natl. Acad. Sci. USA 1997, 94, 3860–3865. [Google Scholar] [CrossRef]
- Courcelle, J.; Crowley, D.J.; Hanawalt, P.C. Recovery of DNA replication in UV-irradiated Escherichia coli requires both excision repair and recF protein function. J. Bacteriol. 1999, 181, 916–922. [Google Scholar] [CrossRef]
- Sharma, R.C.; Sargentini, N.J.; Smith, K.C. New mutation (mmrA1) in Escherichia coli K-12 that affects minimal medium recovery and postreplication repair after UV irradiation. J. Bacteriol. 1983, 154, 743–747. [Google Scholar] [CrossRef]
- Smith, K.C.; Meun, D.H. Repair of radiation-induced damage in Escherichia coli: I. Effect of rec mutations on post-replication repair of damage due to ultraviolet radiation. J. Mol. Biol. 1970, 51, 459–472. [Google Scholar] [CrossRef]
- Ganesan, A.K.; Smith, K.C. The duration of recovery and DNA repair in excision deficient derivatives of Escherichia coli K-12 after ultraviolet irradiation. Mol. Gen. Genet. 1971, 113, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.K. Persistence of pyrimidine dimers during post-replication repair in ultraviolet light-irradiated Escherichia coli K12. J. Mol. Biol. 1974, 87, 103–119. [Google Scholar] [CrossRef]
- Youngs, D.A.; Smith, K.C. Genetic control of multiple pathways of post-replicational repair in uvrB strains of Escherichia coli K-12. J. Bacteriol. 1976, 125, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Barfknecht, T.R.; Smith, K.C. Ultraviolet radiation-induced mutability of isogenic uvrA and uvrB strains of Escherichia coli K-12 W3110. Photochem. Photobiol. 1977, 26, 643–645. [Google Scholar] [CrossRef]
- Sargentini, N.J.; Smith, K.C. Involvement of genes uvrD and recB in separate mutagenic deoxyribonucleic acid repair path-ways in Escherichia coli K-12 uvrB5 and B/r uvrA155. J. Bacteriol. 1980, 143, 212–220. [Google Scholar] [CrossRef]
- Wang, T.V.; Smith, K.C. Effect of recB21, uvrD3, lexA101 and recF143 mutations on ultraviolet radiation sensitivity and genetic recombination in delta uvrB strains of Escherichia coli K-12. Mol. Gen. Genet. 1981, 183, 37–44. [Google Scholar] [CrossRef]
- Wang, T.C.; Smith, K.C. Effects of the ssb-1 and ssb-113 mutations on survival and DNA repair in UV-irradiated delta uvrB strains of Escherichia coli K-12. J. Bacteriol. 1982, 151, 186–192. [Google Scholar] [CrossRef]
- Sharma, R.C.; Barfknecht, T.R.; Smith, K.C. Postreplication repair in uvrA and uvrB strains of escherichia COLI K-12 IS INHIBITED BY RICH GROWTH MEDIUM. Photochem. Photobiol. 1982, 36, 307–311. [Google Scholar] [CrossRef]
- Sargentini, N.J.; Bockrath, R.C.; Smith, K.C. Three mechanisms for ultraviolet radiation mutagenesis in Escherichia coli K-12 uvrB5: Specificity for the production of back and suppressor mutants. Mutat. Res. Mol. Mech. Mutagen. 1982, 106, 217–224. [Google Scholar] [CrossRef]
- Sharma, R.C.; Smith, K.C. Inducible postreplication repair is responsible for minimal medium recovery in UV-irradiated Escherichia coli K-12. Photochem. Photobiol. 1983, 38, 301–303. [Google Scholar] [CrossRef]
- Wang, T.C.; Smith, K.C. Mechanisms for recF-dependent and recB-dependent pathways of postreplication repair in UV-irradiated Escherichia coli uvrB. J. Bacteriol. 1983, 156, 1093–1098. [Google Scholar] [CrossRef]
- Wang, T.V.; Smith, K.C. recF-dependent and recF recB-independent DNA gapfilling repair processes transfer dimercontaining parental strands to daughter strands in Escherichia coli K-12 uvrB. J. Bacteriol. 1984, 158, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-C.V.; Smith, K.C. Role of the umuC gene in postreplication repair in UV-irradiated Escherichia coli K-12 uvrB. Mutat. Res. Repair Rep. 1985, 145, 107–112. [Google Scholar] [CrossRef]
- Sharma, R.C.; Smith, K.C. A minor pathway of postreplication repair in Escherichia coli is independent of the recB, recC and recF genes. Mutat. Res. Repair Rep. 1985, 146, 169–176. [Google Scholar] [CrossRef]
- Sharma, R.C.; Smith, K.C. A mechanism for rich-medium inhibition of the repair of daughter-strand gaps in the deoxyribonucleic acid of UV-irradiated Escherichia coli K12 uvrA. Mutat. Res. 1985, 146, 177–183. [Google Scholar] [PubMed]
- Wang, T.C.; Smith, K.C. Mechanism of sbcB-suppression of the recBC-deficiency in postreplication repair in UV-irradiated Escherichia coli K-12. Mol. Gen. Genet. 1985, 201, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-C.V.; Smith, K.C. Postreplicational formation and repair of DNA double-strand breaks in UV-irradiated Escherichia coli uvrB cells. Mutat. Res. Repair Rep. 1986, 165, 39–44. [Google Scholar] [CrossRef]
- Sargentini, N.J.; Smith, K.C. Role of the radB gene in postreplication repair in UV-irradiated Escherichia coli uvrB. Mutat. Res. Repair Rep. 1986, 166, 17–22. [Google Scholar] [CrossRef]
- Sharma, R.C.; Smith, K.C. Repair of DNA double-strand breaks in UV-irradiated Escherichia coli uvrB recF cells is inhibited by rich growth medium. Mutat. Res. Repair Rep. 1986, 166, 23–28. [Google Scholar] [CrossRef]
- Wang, T.-C.; Smith, K.C. recA (Srf) suppression of recF deficiency in the postreplication repair of UV-irradiated Escherichia coli K-12. J. Bacteriol. 1986, 168, 940–946. [Google Scholar] [CrossRef][Green Version]
- Wang, T.-C.; Smith, K. Different effects of recJ and recN mutations on the postreplication repair of UV-damaged DNA in Escherichia coli K-12. J. Bacteriol. 1988, 170, 2555–2559. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.K.; Seawell, P.C. The effect of lexA and recF mutations on post-replication repair and DNA synthesis in Escherichia coli K-12. Mol. Gen. Genet. 1975, 141, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Pagès, V.; Mazón, G.; Naiman, K.; Philippin, G.; Fuchs, R.P. Monitoring bypass of single replication-blocking lesions by damage avoidance in the Escherichia coli chromosome. Nucleic Acids Res. 2012, 40, 9036–9043. [Google Scholar] [CrossRef] [PubMed]
- Naiman, K.; Philippin, G.; Fuchs, R.P.; Pages, V. Chronology in lesion tolerance gives priority to genetic variability. Proc. Natl. Acad. Sci. USA 2014, 111, 5526–5531. [Google Scholar] [CrossRef]
- Laureti, L.; Demol, J.; Fuchs, R.P.; Pagès, V. Bacterial Proliferation: Keep Dividing and Don’t Mind the Gap. PLoS Genet. 2015, 11, e1005757. [Google Scholar] [CrossRef]
- Naiman, K.; Pagès, V.; Fuchs, R.P. A defect in homologous recombination leads to increased translesion synthesis in E. coli. Nucleic Acids Res. 2016, 44, 7691–7699. [Google Scholar] [CrossRef]
- Laureti, L.; Lee, L.; Philippin, G.; Pagès, V. A non-catalytic role of RecBCD in homology directed gap repair and translesion synthesis. Nucleic Acids Res. 2017, 45, 5877–5886. [Google Scholar] [CrossRef]
- Chrabaszcz, É.; Laureti, L.; Pagès, V. DNA lesions proximity modulates damage tolerance pathways in Escherichia coli. Nucleic Acids Res. 2018, 46, 4004–4012. [Google Scholar] [CrossRef]
- Howard-Flanders, P.; Theriot, L.; Stedeford, J.B. Some Properties of Excision-defective Recombination-deficient Mutants of Escherichia coli K-12. J. Bacteriol. 1969, 97, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Courcelle, J.; Carswell-Crumpton, C.; Hanawalt, P.C. recF and recR are required for the resumption of replication at DNA replication forks in Escherichia coli. Proc. Natl. Acad. Sci. USA 1997, 94, 3714–3719. [Google Scholar] [CrossRef]
- Courcelle, J.; Hanawalt, P.C. RecQ and RecJ process blocked replication forks prior to the resumption of replication in UV-irradiated Escherichia coli. Mol. Gen. Genet. 1999, 262, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Courcelle, J.; Ganesan, A.K.; Hanawalt, P.C. Therefore, what are recombination proteins there for? Bioessays 2001, 23, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Courcelle, J.; Donaldson, J.R.; Chow, K.H.; Courcelle, C.T. DNA Damage-Induced Replication Fork Regression and Processing in Escherichia coli. Science 2003, 299, 1064–1067. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.-H.; Courcelle, J. RecO Acts with RecF and RecR to Protect and Maintain Replication Forks Blocked by UV-induced DNA Damage in Escherichia coli. J. Biol. Chem. 2004, 279, 3492–3496. [Google Scholar] [CrossRef] [PubMed]
- Courcelle, C.T.; Belle, J.J.; Courcelle, J. Nucleotide Excision Repair or Polymerase V-Mediated Lesion Bypass Can Act To Restore UV-Arrested Replication Forks in Escherichia coli. J. Bacteriol. 2005, 187, 6953–6961. [Google Scholar] [CrossRef]
- Courcelle, C.T.; Chow, K.-H.; Casey, A.; Courcelle, J. Nascent DNA processing by RecJ favors lesion repair over translesion synthesis at arrested replication forks in Escherichia coli. Proc. Natl. Acad. Sci. USA 2006, 103, 9154–9159. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.H.; Courcelle, J. RecBCD and RecJ/RecQ initiate DNA degradation on distinct substrates in UV-irradiated Escherichia coli. Radiat. Res. 2007, 168, 499–506. [Google Scholar] [CrossRef]
- Jeiranian, H.A.; Schalow, B.J.; Courcelle, C.T.; Courcelle, J. Fate of the replisome following arrest by UV-induced DNA damage in Escherichia coli. Proc. Natl. Acad. Sci. USA 2013, 110, 11421–11426. [Google Scholar] [CrossRef]
- Wendel, B.M.; Hollingsworth, S.; Courcelle, C.T.; Courcelle, J. UV-induced DNA damage disrupts the coordination between replication initiation, elongation and completion. Genes Cells 2021, 26, 94–108. [Google Scholar] [CrossRef]
- Magner, D.B.; Blankschien, M.D.; Lee, J.A.; Pennington, J.M.; Lupski, J.R.; Rosenberg, S.M. RecQ Promotes Toxic Recombination in Cells Lacking Recombination Intermediate-Removal Proteins. Mol. Cell 2007, 26, 273–286. [Google Scholar] [CrossRef]
- Fonville, N.C.; Blankschien, M.D.; Magner, D.B.; Rosenberg, S.M. RecQ-dependent death-by-recombination in cells lacking RecG and UvrD. DNA Repair 2010, 9, 403–413. [Google Scholar] [CrossRef][Green Version]
- Kusano, K.; Nakayama, K.; Nakayama, H. Plasmid-mediated lethality and plasmid multimer formation in an Escherichia coli recBC sbcBC mutant. Involvement of RecF recombination pathway genes. J. Mol. Biol. 1989, 209, 623–634. [Google Scholar] [CrossRef]
- Hanada, K.; Iwasaki, M.; Ihashi, S.; Ikeda, H. UvrA and UvrB suppress illegitimate recombination: Synergistic action with RecQ helicase. Proc. Natl. Acad. Sci. USA 2000, 97, 5989–5994. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Nakayama, K.; Nakayama, R.; Irino, N.; Nakayama, Y.; Hanawalt, P.C. Isolation and genetic characterization of a thymineless death-resistant mutant of Escherichia coli K12: Identification of a new mutation (recQ1) that blocks the RecF recombination pathway. Mol. Gen. Genet. 1984, 195, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Ream, L.W.; Margossian, L.; Clark, A.J.; Hansen, F.G.; von Meyenburg, K. Genetic and physical mapping of recF in Escherichia coli K-12. Mol. Gen. Genet. 1980, 180, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Perez-Roger, I.; Garcia-Sogo, M.; Navarro-Avino, J.P.; Lopez-Acedo, C.; Macian, F.; Armengod, M.E. Positive and negative regulatory elements in the dnaA-dnaN-recF operon of Escherichia coli. Biochimie 1991, 73, 329–334. [Google Scholar] [CrossRef]
- Flower, A.M.; McHenry, C.S. Transcriptional organization of the Escherichia coli dnaX gene. J. Mol. Biol. 1991, 220, 649–658. [Google Scholar] [CrossRef]
- Luisi-DeLuca, C.; Kolodner, R. Purification and characterization of the Escherichia coli RecO protein. Renaturation of complementary single-stranded DNA molecules catalyzed by the RecO protein. J. Mol. Biol. 1994, 236, 124–138. [Google Scholar] [CrossRef]
- Britton, R.A.; Powell, B.S.; Court, D.L.; Lupski, J.R. Characterization of mutations affecting the Escherichia coli essential GTPase era that suppress two temperature-sensitive dnaG alleles. J. Bacteriol. 1997, 179, 4575–4582. [Google Scholar] [CrossRef]
- Britton, R.A.; Lupski, J.R. Isolation and Characterization of Suppressors of Two Escherichia coli dnaG Mutations, dnaG2903 and parB. Genetics 1997, 145, 867–875. [Google Scholar] [CrossRef]
- Britton, R.A.; Powell, B.S.; Dasgupta, S.; Sun, Q.; Margolin, W.; Lupski, J.R.; Court, D.L. Cell cycle arrest in Era GTPase mutants: A potential growth rate-regulated checkpoint in Escherichia coli. Mol. Microbiol. 1998, 27, 739–750. [Google Scholar] [CrossRef] [PubMed]
- McInerney, P.; O’donnell, M. Replisome Fate upon Encountering a Leading Strand Block and Clearance from DNA by Recombination Proteins. J. Biol. Chem. 2007, 282, 25903–25916. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, K.; Katayama, T.; Iwai, S.; Hidaka, M.; Horiuchi, T.; Maki, H. Fate of DNA replication fork encountering a single DNA lesion during oriC plasmid DNA replication in vitro. Genes Cells 2003, 8, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Pages, V.; Fuchs, R.P. Uncoupling of Leading- and Lagging-Strand DNA Replication During Lesion Bypass in Vivo. Science 2003, 300, 1300–1303. [Google Scholar] [CrossRef]
- Svoboda, D.L.; Vos, J.M. Differential replication of a single, UV-induced lesion in the leading or lagging strand by a human cell extract: Fork uncoupling or gap formation. Proc. Natl. Acad. Sci. USA 1995, 92, 11975–11979. [Google Scholar] [CrossRef]
- Svoboda, D.L.; Briley, L.P.; Vos, J.M. Defective bypass replication of a leading strand cyclobutane thymine dimer in xeroderma pigmentosum variant cell extracts. Cancer Res. 1998, 58, 2445–2448. [Google Scholar]
- Setlow, R.B.; Swenson, P.A.; Carrier, W.L. Thymine dimers and inhibition of DNA synthesis by UL-traviolet irradiation of cells. Science 1963, 142, 1464–1466. [Google Scholar] [CrossRef]
- Chan, G.L.; Doetsch, P.W.; Haseltine, W.A. Cyclobutane pyrimidine dimers and (6-4) photoproducts block polymerization by DNA polymerase I. Biochemistry 1985, 24, 5723–5728. [Google Scholar] [CrossRef]
- Belle, J.J.; Casey, A.; Courcelle, C.T.; Courcelle, J. Inactivation of the DnaB Helicase Leads to the Collapse and Degradation of the Replication Fork: A Comparison to UV-Induced Arrest. J. Bacteriol. 2007, 189, 5452–5462. [Google Scholar] [CrossRef][Green Version]
- Donaldson, J.R.; Courcelle, C.T.; Courcelle, J. RuvABC is required to resolve holliday junctions that accumulate following replication on damaged templates in Escherichia coli. J. Biol. Chem. 2006, 281, 28811–28821. [Google Scholar] [CrossRef]
- Lovett, S.; Kolodner, R.D. Identification and purification of a single-stranded-DNA-specific exonuclease encoded by the recJ gene of Escherichia coli. Proc. Natl. Acad. Sci. USA 1989, 86, 2627–2631. [Google Scholar] [CrossRef] [PubMed]
- Umezu, K.; Nakayama, K.; Nakayama, H. Escherichia coli RecQ protein is a DNA helicase. Proc. Natl. Acad. Sci. USA 1990, 87, 5363–5367. [Google Scholar] [CrossRef] [PubMed]
- Bichara, M.; Pinet, I.; Lambert, I.B.; Fuchs, R.P. RecA-mediated excision repair: A novel mechanism for repairing DNA lesions at sites of arrested DNA synthesis. Mol. Microbiol. 2007, 65, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Bichara, M.; Fuchs, R.P.; Cordonnier, A.; Lambert, I.B. Preferential post-replication repair of DNA lesions situated on the leading strand of plasmids in Escherichia coli. Mol. Microbiol. 2009, 71, 305–314. [Google Scholar] [CrossRef]
- Postow, L.; Ullsperger, C.; Keller, R.W.; Bustamante, C.; Vologodskii, A.V.; Cozzarelli, N.R. Positive Torsional Strain Causes the Formation of a Four-way Junction at Replication Forks. J. Biol. Chem. 2001, 276, 2790–2796. [Google Scholar] [CrossRef]
- Fierro-Fernandez, M.; Hernandez, P.; Krimer, D.B.; Schvartzman, J.B. Replication fork reversal occurs spontaneously after digestion but is constrained in supercoiled domains. J. Biol. Chem. 2007, 282, 18190–18196. [Google Scholar] [CrossRef]
- McGlynn, P.; Lloyd, R.G. Action of RuvAB at Replication Fork Structures. J. Biol. Chem. 2001, 276, 41938–41944. [Google Scholar] [CrossRef]
- McGlynn, P.; Lloyd, R.G.; Marians, K.J. Formation of Holliday junctions by regression of nascent DNA in intermediates containing stalled replication forks: RecG stimulates regression even when the DNA is negatively supercoiled. Proc. Natl. Acad. Sci. USA 2001, 98, 8235–8240. [Google Scholar] [CrossRef]
- Baharoglu, Z.; Petranovic, M.; Flores, M.-J.; Michel, B. RuvAB is essential for replication forks reversal in certain replication mutants. EMBO J. 2006, 25, 596–604. [Google Scholar] [CrossRef]
- Le Masson, M.; Baharoglu, Z.; Michel, B. RuvA and ruvB mutants specifically impaired for replication fork reversal. Mol. Microbiol. 2008, 70, 537–548. [Google Scholar] [CrossRef]
- Donaldson, J.R.; Courcelle, C.T.; Courcelle, J. RuvAB and RecG Are Not Essential for the Recovery of DNA Synthesis Following UV-Induced DNA Damage in Escherichia coli. Genetics 2004, 166, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, J.E.; Cox, M.M. On RecA protein-mediated homologous alignment of two DNA molecules. Three strands versus four strands. J. Biol. Chem. 1990, 265, 10164–10171. [Google Scholar] [CrossRef]
- Cox, M.M.; Lehman, I.R. Directionality and polarity in recA protein-promoted branch migration. Proc. Natl. Acad. Sci. USA 1981, 78, 6018–6022. [Google Scholar] [CrossRef] [PubMed]
- Shan, Q.; Cox, M.M. RecA Filament Dynamics during DNA Strand Exchange Reactions. J. Biol. Chem. 1997, 272, 11063–11073. [Google Scholar] [CrossRef] [PubMed]
- Shana, Q.; Bork, J.M.; Webb, B.L.; Inman, R.B.; Cox, M.M. RecA protein filaments: End-dependent dissociation from ssDNA and stabilization by RecO and RecR proteins. J. Mol. Biol. 1997, 265, 519–540. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.M. Motoring along with the bacterial RecA protein. Nat. Rev. Mol. Cell Biol. 2007, 8, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Hishida, T.; Han, Y.-W.; Shibata, T.; Kubota, Y.; Ishino, Y.; Iwasaki, H.; Shinagawa, H. Role of the Escherichia coli RecQ DNA helicase in SOS signaling and genome stabilization at stalled replication forks. Genes Dev. 2004, 18, 1886–1897. [Google Scholar] [CrossRef]
- Setlow, R.B.; Carrier, W.L. The disappearance of thymine dimers from DNA: An error-correcting mechanism. Proc. Natl. Acad. Sci. USA 1964, 51, 226–231. [Google Scholar] [CrossRef]
- Sancar, A.; Rupp, W. A novel repair enzyme: UVRABC excision nuclease of Escherichia coli cuts a DNA strand on both sides of the damaged region. Cell 1983, 33, 249–260. [Google Scholar] [CrossRef]
- Husain, I.; Van Houten, B.; Thomas, D.C.; Abdel-Monem, M.; Sancar, A. Effect of DNA polymerase I and DNA helicase II on the turnover rate of UvrABC excision nuclease. Proc. Natl. Acad. Sci. USA 1985, 82, 6774–6778. [Google Scholar] [CrossRef]
- Caron, P.R.; Kushner, S.R.; Grossman, L. Involvement of helicase II (uvrD gene product) and DNA polymerase I in excision mediated by the uvrABC protein complex. Proc. Natl. Acad. Sci. USA 1985, 82, 4925–4929. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, B. Nucleotide excision repair in Escherichia coli. Microbiol. Rev. 1990, 54, 18–51. [Google Scholar] [CrossRef] [PubMed]
- Howard-Flanders, P.; Boyce, R.P.; Theriot, L. Three loci in Escherichia coli K-12 that control the excision of pyrimidine dimers and certain other mutagen products from DNA. Genetics 1966, 53, 1119–1136. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Cooper, P.K.; Hanawalt, P.C. DNA Repair A manual of Research ProceduresMeasurement of Repair Repli-Cation by Equilibrium Sedimentation; Friedberg, E.C., Hanawalt, P.C., Eds.; 1B; Marcel Dekker Inc.: New York, NY, USA, 1981. [Google Scholar]
- Courcelle, C.T.; Courcelle, J. Monitoring DNA Replication Following UV-Induced Damage in Escherichia coli. Methods Enzymol. 2006, 409, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, C.J.; Upton, A.L.; Lloyd, R.G. Maintaining replication fork integrity in UV-irradiated Escherichia coli cells. DNA Repair 2008, 7, 1589–1602. [Google Scholar] [CrossRef] [PubMed]
- Witkin, E.M.; Roegner-Maniscalco, V.; Sweasy, J.B.; McCall, J.O. Recovery from ultraviolet light-induced inhibition of DNA synthesis requires umuDC gene products in recA718 mutant strains but not in recA+ strains of Escherichia coli. Proc. Natl. Acad. Sci. USA 1987, 84, 6805–6809. [Google Scholar] [CrossRef] [PubMed]
- Friedman, K.L.; Brewer, B.J. Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol. 1995, 262, 613–627. [Google Scholar] [CrossRef]
- Kolodner, R.; Fishel, R.A.; Howard, M. Genetic recombination of bacterial plasmid DNA: Effect of RecF pathway mutations on plasmid recombination in Escherichia coli. J. Bacteriol. 1985, 163, 1060–1066. [Google Scholar] [CrossRef]
- Mahdi, A.A.; Lloyd, R.G. Identification of the recR locus of Escherichia coli K-12 and analysis of its role in recombination and DNA repair. Mol. Gen. Genet. 1989, 216, 503–510. [Google Scholar] [CrossRef]
- Thoms, B.; Wackernagel, W. Regulatory role of recF in the SOS response of Escherichia coli: Impaired induction of SOS genes by UV irradiation and nalidixic acid in a recF mutant. J. Bacteriol. 1987, 169, 1731–1736. [Google Scholar] [CrossRef]
- Hegde, S.; Sandler, S.J.; Clark, A.J.; Madiraju, M.V.V.S. RecO and recR mutations delay induction of the SOS response in Escherichia coli. Mol. Gen. Genet. 1995, 246, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Whitby, M.; Lloyd, R.G. Altered SOS induction associated with mutations in recF, recO and recR. Mol. Gen. Genet. 1995, 246, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Brent, R.; Ptashne, M. The lexA gene product represses its own promoter. Proc. Natl. Acad. Sci. USA 1980, 77, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- Brent, R.; Ptashne, M. Mechanism of action of the lexA gene product. Proc. Natl. Acad. Sci. USA 1981, 78, 4204–4208. [Google Scholar] [CrossRef]
- Kenyon, C.J.; Brent, R.; Ptashne, M.; Walker, G.C. Regulation of damage-inducible genes in Escherichia coli. J. Mol. Biol. 1982, 160, 445–457. [Google Scholar] [CrossRef]
- Little, J.W.; Mount, D.W.; Yanisch-Perron, C.R. Purified lexA protein is a repressor of the recA and lexA genes. Proc. Natl. Acad. Sci. USA 1981, 78, 4199–4203. [Google Scholar] [CrossRef]
- Sassanfar, M.; Roberts, J.W. Nature of the SOS-inducing signal in Escherichia coli: The involvement of DNA replication. J. Mol. Biol. 1990, 212, 79–96. [Google Scholar] [CrossRef]
- Umezu, K.; Chi, N.W.; Kolodner, R.D. Biochemical interaction of the Escherichia coli RecF, RecO, and RecR proteins with RecA protein and single-stranded DNA binding protein. Proc. Natl. Acad. Sci. USA 1993, 90, 3875–3879. [Google Scholar] [CrossRef]
- Umezu, K.; Kolodner, R.D. Protein interactions in genetic recombination in Escherichia coli. Interactions involving RecO and RecR overcome the inhibition of RecA by single-stranded DNA-binding protein. J. Biol. Chem. 1994, 269, 30005–30013. [Google Scholar] [CrossRef]
- Webb, B.L.; Cox, M.M.; Inman, R.B. An Interaction between the Escherichia coli RecF and RecR Proteins Dependent on ATP and Double-stranded DNA. J. Biol. Chem. 1995, 270, 31397–31404. [Google Scholar] [CrossRef]
- Webb, B.L.; Cox, M.M.; Inman, R.B. Recombinational DNA repair: The RecF and RecR proteins limit the extension of RecA filaments beyond single-strand DNA gaps. Cell 1997, 91, 347–356. [Google Scholar] [CrossRef]
- Henrikus, S.S.; Henry, C.; Ghodke, H.; Wood, E.A.; Mbele, N.; Saxena, R.; Basu, U.; van Oijen, A.; Cox, M.M.; Robinson, A. RecFOR epistasis group: RecF and RecO have distinct localizations and functions in Escherichia coli. Nucleic Acids Res. 2019, 47, 2946–2965. [Google Scholar] [CrossRef] [PubMed]
- Skretas, G.; Georgiou, G. Simple Genetic Selection Protocol for Isolation of Overexpressed Genes That Enhance Accumulation of Membrane-Integrated Human G Protein-Coupled Receptors in Escherichia coli. Appl. Environ. Microbiol. 2010, 76, 5852–5859. [Google Scholar] [CrossRef] [PubMed]
- Opperman, T.; Murli, S.; Smith, B.T.; Walker, G.C. A model for a umuDC-dependent prokaryotic DNA damage checkpoint. Proc. Natl. Acad. Sci. USA 1999, 96, 9218–9223. [Google Scholar] [CrossRef]
- Ferentz, A.E.; Walker, G.C.; Wagner, G. Converting a DNA damage checkpoint effector (UmuD2C) into a lesion bypass polymerase (UmuD’2C). EMBO J. 2001, 20, 4287–4298. [Google Scholar] [CrossRef]
- Ollivierre, J.N.; Fang, J.; Beuning, P.J. The Roles of UmuD in Regulating Mutagenesis. J. Nucleic Acids 2010, 2010, 947680. [Google Scholar] [CrossRef]
- Ollivierre, J.N.; Sikora, J.L.; Beuning, P.J. Dimer exchange and cleavage specificity of the DNA damage response protein UmuD. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2013, 1834, 611–620. [Google Scholar] [CrossRef]
- Kato, T.; Shinoura, Y. Isolation and characterization of mutants of Escherichia coli deficient in induction of mutations by ultraviolet light. Mol. Gen. Genet. 1977, 156, 121–131. [Google Scholar] [CrossRef]
- Bagg, A.; Kenyon, C.J.; Walker, G.C. Inducibility of a gene product required for UV and chemical mutagenesis in Escherichia coli. Proc. Natl. Acad. Sci. USA 1981, 78, 5749–5753. [Google Scholar] [CrossRef]
- Reuven, N.B.; Arad, G.; Maor-Shoshani, A.; Livneh, Z. The mutagenesis protein UmuC is a DNA polymerase activated by UmuD’, RecA, and SSB and is specialized for translesion replication. J. Biol. Chem. 1999, 274, 31763–31766. [Google Scholar] [CrossRef]
- Tang, M.; Shen, X.; Frank, E.G.; O’Donnell, M.; Woodgate, R.; Goodman, M.F. UmuD’(2)C is an error-prone DNA polymerase, Escherichia coli pol V. Proc. Natl. Acad. Sci. USA 1999, 96, 8919–8924. [Google Scholar] [CrossRef] [PubMed]
- Sommer, S.; Bailone, A.; Devoret, R. The appearance of the UmuD’C protein complex in Escherichia coli switches repair from homologous recombination to SOS mutagenesis. Mol. Microbiol. 1993, 10, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Shinagawa, H.; Iwasaki, H.; Kato, T.; Nakata, A. RecA protein-dependent cleavage of UmuD protein and SOS mutagenesis. Proc. Natl. Acad. Sci. USA 1988, 85, 1806–1810. [Google Scholar] [CrossRef] [PubMed]
- Nohmi, T.; Battista, J.R.; Dodson, L.A.; Walker, G.C. RecA-mediated cleavage activates UmuD for mutagenesis: Mechanistic relationship between transcriptional derepression and posttranslational activation. Proc. Natl. Acad. Sci. USA 1988, 85, 1816–1820. [Google Scholar] [CrossRef] [PubMed]
- Davey, S.; Han, C.S.; Ramer, S.A.; Klassen, J.C.; Jacobson, A.; Eisenberger, A.; Hopkins, K.M.; Lieberman, H.B.; Freyer, G.A. Fission Yeast rad12 + Regulates Cell Cycle Checkpoint Control and Is Homologous to the Bloom’s Syndrome Disease Gene. Mol. Cell. Biol. 1998, 18, 2721–2728. [Google Scholar] [CrossRef]
- Ellis, N.A.; Groden, J.; Ye, T.-Z.; Straughen, J.; Lennon, D.J.; Ciocci, S.; Proytcheva, M.; German, J. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 1995, 83, 655–666. [Google Scholar] [CrossRef]
- Gray, M.D.; Shen, J.C.; Kamath-Loeb, A.S.; Blank, A.; Sopher, B.L.; Martin, G.M.; Oshima, J.; Loeb, L.A. The Werner syndrome protein is a DNA helicase. Nat. Genet. 1997, 17, 100–103. [Google Scholar] [CrossRef]
- Stewart, E.; Chapman, C.R.; Al-Khodairy, F.; Carr, A.M.; Enoch, T. rqh1+, a fission yeast gene related to the Bloom’s and Werner’s syndrome genes, is required for reversible S phase arrest. EMBO J. 1997, 16, 2682–2692. [Google Scholar] [CrossRef]
- Yan, Z.; Xue, C.; Kumar, S.; Crickard, J.B.; Yu, Y.; Wang, W.; Pham, N.; Li, Y.; Niu, H.; Sung, P.; et al. Rad52 Restrains Resection at DNA Double-Strand Break Ends in Yeast. Mol. Cell 2019, 76, 699–711.e6. [Google Scholar] [CrossRef]
- Kitao, S.; Shimamoto, A.; Goto, M.; Miller, R.W.; Smithson, W.A.; Lindor, N.M.; Furuichi, Y. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat. Genet. 1999, 22, 82–84. [Google Scholar] [CrossRef]
- Murray, J.M.; Lindsay, H.D.; Munday, C.A.; Carr, A.M. Role of Schizosaccharomyces pombe RecQ homolog, recombination, and checkpoint genes in UV damage tolerance. Mol. Cell. Biol. 1997, 17, 6868–6875. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Kato, J.; Shimamoto, A.; Goto, M.; Furuichi, Y.; Ikeda, H. Bloom’s and Werner’s syndrome genes suppress hyperrecombination in yeast sgs1 mutant: Implication for genomic instability in human diseases. Proc. Natl. Acad. Sci. USA 1998, 95, 8733–8738. [Google Scholar] [CrossRef] [PubMed]
- Watt, P.M.; Hickson, I.D.; Borts, R.H.; Louis, E.J. SGS1, a Homologue of the Bloom’s and Werner’s Syndrome Genes, Is Required for Maintenance of Genome Stability in Saccharomyces cerevisiae. Genetics 1996, 144, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Gangloff, S.; McDonald, J.P.; Bendixen, C.; Arthur, L.; Rothstein, R. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: A potential eukaryotic reverse gyrase. Mol. Cell. Biol. 1994, 14, 8391–8398. [Google Scholar]
- Puranam, K.L.; Blackshear, P.J. Cloning and characterization of RECQL, a potential human homologue of the Escherichia coli DNA helicase RecQ. J. Biol. Chem. 1994, 269, 29838–29845. [Google Scholar] [CrossRef]
- Cooper, P.K.; Hanawalt, P.C. Heterogeneity of patch size in repair replicated DNA in Escherichia coli. J. Mol. Biol. 1972, 67, 1–10. [Google Scholar] [CrossRef]
- Cooper, P.K.; Hanawalt, P.C. Role of DNA Polymerase I and the rec System in Excision-Repair in Escherichia coli. Proc. Natl. Acad. Sci. USA 1972, 69, 1156–1160. [Google Scholar] [CrossRef]
- Cooper, P.K. Characterization of long patch excision repair of DNA in ultraviolet-irradiated Escherichia coli: An inducible function under reclex control. Mol. Gen. Genet. 1982, 185, 189–197. [Google Scholar] [CrossRef]
- Salaj-Smic, E.; Petranović, D.; Petranović, M.; Trgovcević, Z. Relative roles of uvrA and recA genes in the recovery of Escherichia coli and phage lambda after ultraviolet irradiation. Radiat. Res. 1980, 83, 323–329. [Google Scholar] [CrossRef]
- Rudolph, C.J.; Upton, A.L.; Lloyd, R.G. Replication fork stalling and cell cycle arrest in UV-irradiated Escherichia coli. Genes Dev. 2007, 21, 668–681. [Google Scholar] [CrossRef]
- Rothman, R.H.; Clark, A.J. The dependence of postreplication repair on uvrB in a recF mutant of Escherichia coli K-12. Mol. Gen. Genet. 1977, 155, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.E.; Daune, M.P.; Fuchs, R.P. Repair and mutagenesis of plasmid DNA modified by ultraviolet irradiation or N-acetoxy-N-2-acetylaminofluorene. Proc. Natl. Acad. Sci. USA 1982, 79, 4133–4137. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, R.; Seeberg, E. pBR322 plasmid DNA modified with 2-acetylaminofluorene derivatives: Transforming activity and in vitro strand cleavage by the Escherichia coli uvrABC endonuclease. EMBO J. 1984, 3, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.; Ehrlich, S.; Uzest, M. DNA double-strand breaks caused by replication arrest. EMBO J. 1997, 16, 430–438. [Google Scholar] [CrossRef]
- Seigneur, M.; Bidnenko, V.; Ehrlich, S.; Michel, B. RuvAB Acts at Arrested Replication Forks. Cell 1998, 95, 419–430. [Google Scholar] [CrossRef]
- Michel, B. Replication fork arrest and DNA recombination. Trends Biochem. Sci. 2000, 25, 173–178. [Google Scholar] [CrossRef]
- Michel, B.; Flores, M.J.; Viguera, E.; Grompone, G.; Seigneur, M.; Bidnenko, V. Rescue of arrested replication forks by homologous recombination. Proc. Natl. Acad. Sci. USA 2001, 98, 8181–8188. [Google Scholar] [CrossRef]
- McGlynn, P.; Lloyd, R.G. Rescue of stalled replication forks by RecG: Simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc. Natl. Acad. Sci. USA 2001, 98, 8227–8234. [Google Scholar] [CrossRef]
- Gregg, A.V.; McGlynn, P.; Jaktaji, R.P.; Lloyd, R.G. Direct Rescue of Stalled DNA Replication Forks via the Combined Action of PriA and RecG Helicase Activities. Mol. Cell 2002, 9, 241–251. [Google Scholar] [CrossRef]
- McGlynn, P.; Lloyd, R.G. Genome stability and the processing of damaged replication forks by RecG. Trends Genet. 2002, 18, 413–419. [Google Scholar] [CrossRef]
- McGlynn, P.; Lloyd, R.G. Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell Biol. 2002, 3, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Dillingham, M.S.; Kowalczykowski, S.C. A Step Backward in Advancing DNA Replication: Rescue of Stalled Replication Forks by RecG. Mol. Cell 2001, 8, 734–736. [Google Scholar] [CrossRef]
- Ona, K.R.; Courcelle, C.T.; Courcelle, J. Nucleotide excision repair is a predominant mechanism for processing nitrofurazone-induced DNA damage in Escherichia coli. J. Bacteriol. 2009, 191, 4959–4965. [Google Scholar] [CrossRef]
- Wendel, B.; Courcelle, C.T.; Courcelle, J. Completion of DNA replication in Escherichia coli. Proc. Natl. Acad. Sci. USA 2014, 111, 16454–16459. [Google Scholar] [CrossRef] [PubMed]
- Khidhir, M.A.; Casaregola, S.; Holland, I.B. Mechanism of transient inhibition of DNA synthesis in ultraviolet-irradiated E. coli: Inhibition is independent of recA whilst recovery requires RecA protein itself and an additional, inducible SOS function. Mol. Gen. Genet. 1985, 199, 133–140. [Google Scholar] [CrossRef]
- Kuzminov, A. Collapse and repair of replication forks in Escherichia coli. Mol. Microbiol. 1995, 16, 373–384. [Google Scholar] [CrossRef]
- Kuzminov, A. Instability of inhibited replication forks in E. coli. BioEssays 1995, 17, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.; Boubakri, H.; Baharoglu, Z.; Lemasson, M.; Lestini, R. Recombination proteins and rescue of arrested replication forks. DNA Repair 2007, 6, 967–980. [Google Scholar] [CrossRef]
- Flores, M.J.; Ehrlich, S.D.; Michel, B. Primosome assembly requirement for replication restart in the Escherichia coli holDG10 replication mutant. Mol. Microbiol. 2002, 44, 783–792. [Google Scholar] [CrossRef]
- Grompone, G.; Seigneur, M.; Ehrlich, S.D.; Michel, B. Replication fork reversal in DNA polymerase III mutants of Escherichia coli: A role for the beta clamp. Mol. Microbiol. 2002, 44, 1331–1339. [Google Scholar] [CrossRef]
- Grompone, G.; Ehrlich, D.; Michel, B. Cells defective for replication restart undergo replication fork reversal. EMBO Rep. 2004, 5, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Grompone, G.; Sanchez, N.; Dusko Ehrlich, S.; Michel, B. Requirement for RecFOR-mediated recombination in priA mutant. Mol. Microbiol. 2004, 52, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Seigneur, M.; Ehrlich, S.D.; Michel, B. RecD sbcB sbcD Mutants Are Deficient in Recombinational Repair of UV Lesions by RecBC. J. Bacteriol. 1999, 181, 6220–6221. [Google Scholar] [CrossRef]
- Rothstein, R.; Michel, B.; Gangloff, S. Replication fork pausing and recombination or “gimme a break”. Genes Dev. 2000, 14, 1–10. [Google Scholar] [CrossRef]
- Seigneur, M.; Ehrlich, S.D.; Michel, B. RuvABC-dependent double-strand breaks in dnaBts mutants require recA. Mol. Microbiol. 2000, 38, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Uzest, M.; Ehrlich, S.D.; Michel, B. Lethality of rep recB and rep recC double mutants of Escherichia coli. Mol. Microbiol. 1995, 17, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, C.J.; Upton, A.L.; Stockum, A.; Nieduszynski, C.; Lloyd, R.G. Avoiding chromosome pathology when replication forks collide. Nature 2013, 500, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Courcelle, J.; Wendel, B.M.; Livingstone, D.D.; Courcelle, C.T. RecBCD is required to complete chromosomal replication: Implications for double-strand break frequencies and repair mechanisms. DNA Repair 2015, 32, 86–95. [Google Scholar] [CrossRef][Green Version]
- Flores, M.-J.; Bierne, H.; Ehrlich, S.D.; Michel, B. Impairment of lagging strand synthesis triggers the formation of a RuvABC substrate at replication forks. EMBO J. 2001, 20, 619–629. [Google Scholar] [CrossRef]
- Dimude, J.U.; Midgley-Smith, S.L.; Stein, M.; Rudolph, C.J. Replication Termination: Containing Fork Fusion-Mediated Pathologies in Escherichia coli. Genes 2016, 7, 40. [Google Scholar] [CrossRef]
- Midgley-Smith, S.L.; Dimude, J.U.; Rudolph, C.J. A role for 3′ exonucleases at the final stages of chromosome duplication in Escherichia coli. Nucleic Acids Res. 2018, 47, 1847–1860. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.; Maccabee, M.; Hatahet, Z.; Courcelle, J.; Bockrath, R.; Ide, H.; Wallace, S.S. Thymine ring saturation and fragmentation products: Lesion bypass, misinsertion and implications for mutagenesis. Mutat. Res. Toxicol. 1993, 299, 147–156. [Google Scholar] [CrossRef]
- Wendel, B.M.; Cole, J.M.; Courcelle, C.T.; Courcelle, J. SbcC-SbcD and ExoI process convergent forks to complete chromosome replication. Proc. Natl. Acad. Sci. USA 2018, 115, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.A.; Wendel, B.; Weber, E.A.; Courcelle, C.T.; Courcelle, J. RecBCD, SbcCD and ExoI process a substrate created by convergent replisomes to complete DNA replication. Mol. Microbiol. 2019, 111, 1638–1651. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef]
- Siaud, N.; Barbera, M.A.; Egashira, A.; Lam, I.; Christ, N.; Schlacher, K.; Xia, B.; Jasin, M. Plasticity of BRCA2 function in homologous recombination: Genetic interactions of the PALB2 and DNA binding domains. PLoS Genet. 2011, 7, e1002409. [Google Scholar] [CrossRef]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef]
- Lemaçon, D.; Jackson, J.; Quinet, A.; Brickner, J.R.; Li, S.; Yazinski, S.; You, Z.; Ira, G.; Zou, L.; Mosammaparast, N.; et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017, 8, 860. [Google Scholar] [CrossRef]
- Hambarde, S.; Tsai, C.-L.; Pandita, R.K.; Bacolla, A.; Maitra, A.; Charaka, V.; Hunt, C.R.; Kumar, R.; Limbo, O.; Le Meur, R.; et al. EXO5-DNA structure and BLM interactions direct DNA resection critical for ATR-dependent replication restart. Mol. Cell 2021, 81, 2989–3006.e9. [Google Scholar] [CrossRef]
- Somyajit, K.; Saxena, S.; Babu, S.; Mishra, A.; Nagaraju, G. Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart. Nucleic Acids Res. 2015, 43, 9835–9855. [Google Scholar] [CrossRef]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Mojumdar, A. Mutations in conserved functional domains of human RecQ helicases are associated with diseases and cancer: A review. Biophys. Chem. 2020, 265, 106433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lian, H.; Chen, K.; Pang, Y.; Chen, M.; Huang, B.; Zhu, L.; Xu, S.; Liu, M.; Zhong, C. RECQ1 Promotes Stress Resistance and DNA Replication Progression Through PARP1 Signaling Pathway in Glioblastoma. Front. Cell Dev. Biol. 2021, 9, 714868. [Google Scholar] [CrossRef]
- Blank, A.; Bobola, M.S.; Gold, B.; Varadarajan, S.; Kolstoe, D.D.; Meade, E.H.; Rabinovitch, P.S.; Loeb, L.A.; Silber, J.R. The Werner syndrome protein confers resistance to the DNA lesions N3-methyladenine and O6-methylguanine: Implications for WRN function. DNA Repair 2004, 3, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Biswas, K.; Sommers, J.A.; Thompson, H.; Awate, S.; Nicolae, C.M.; Thakar, T.; Moldovan, G.-L.; Shoemaker, R.H.; Sharan, S.K.; et al. WRN helicase safeguards deprotected replication forks in BRCA2-mutated cancer cells. Nat. Commun. 2021, 12, 6561. [Google Scholar] [CrossRef] [PubMed]
- Chaganti, R.S.; Schonberg, S.; German, J. A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc. Natl. Acad. Sci. USA 1974, 71, 4508–4512. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Courcelle, J.; Worley, T.K.; Courcelle, C.T. Recombination Mediator Proteins: Misnomers That Are Key to Understanding the Genomic Instabilities in Cancer. Genes 2022, 13, 437. https://doi.org/10.3390/genes13030437
Courcelle J, Worley TK, Courcelle CT. Recombination Mediator Proteins: Misnomers That Are Key to Understanding the Genomic Instabilities in Cancer. Genes. 2022; 13(3):437. https://doi.org/10.3390/genes13030437
Chicago/Turabian StyleCourcelle, Justin, Travis K. Worley, and Charmain T. Courcelle. 2022. "Recombination Mediator Proteins: Misnomers That Are Key to Understanding the Genomic Instabilities in Cancer" Genes 13, no. 3: 437. https://doi.org/10.3390/genes13030437
APA StyleCourcelle, J., Worley, T. K., & Courcelle, C. T. (2022). Recombination Mediator Proteins: Misnomers That Are Key to Understanding the Genomic Instabilities in Cancer. Genes, 13(3), 437. https://doi.org/10.3390/genes13030437