
Following Excitation/Inhibition Ratio Homeostasis from Synapse to EEG in Monogenetic Neurodevelopmental Disorders

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
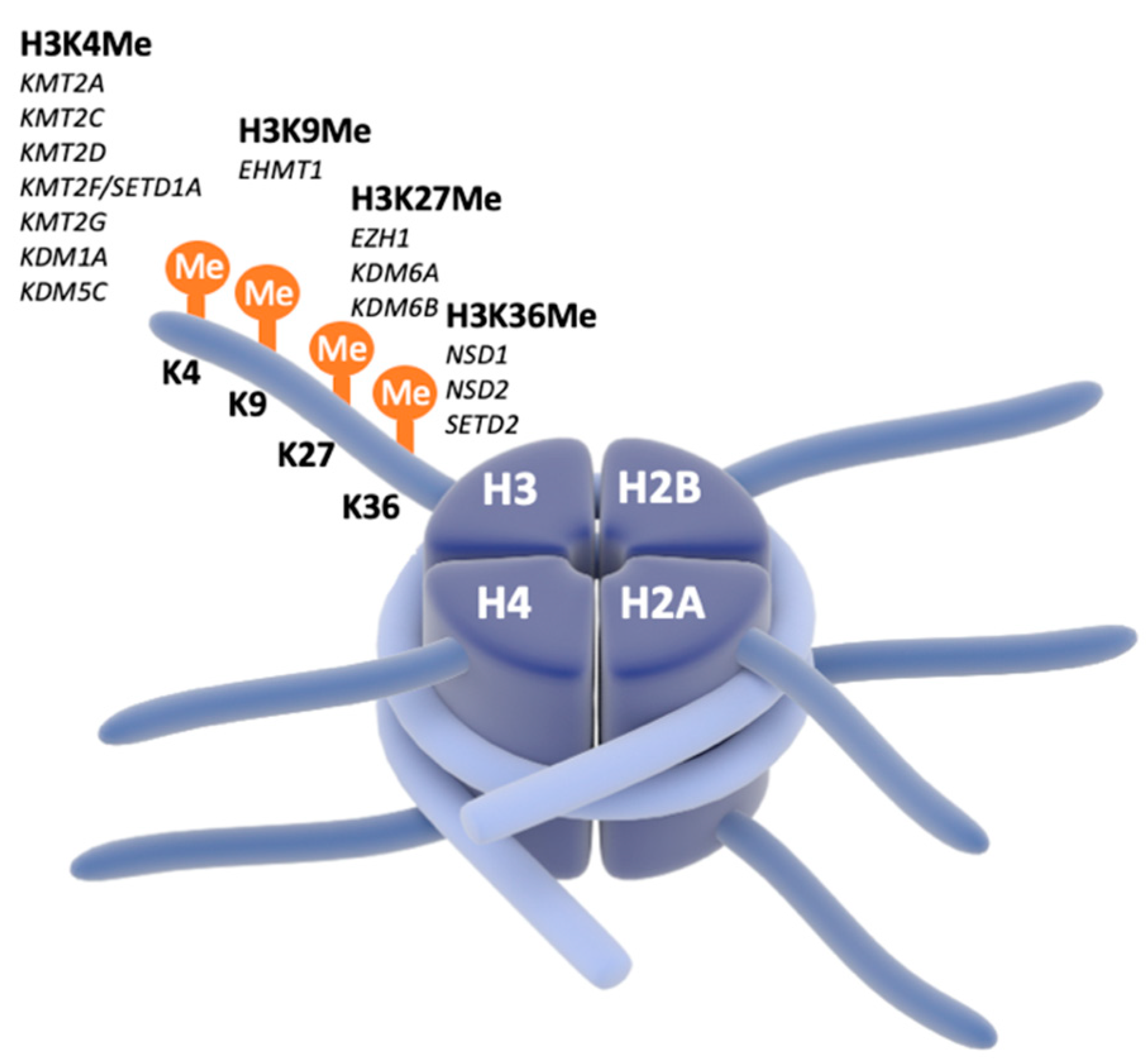
2. Molecular and Physiological Heterogeneity of E/I Ratio Homeostasis in NDDs
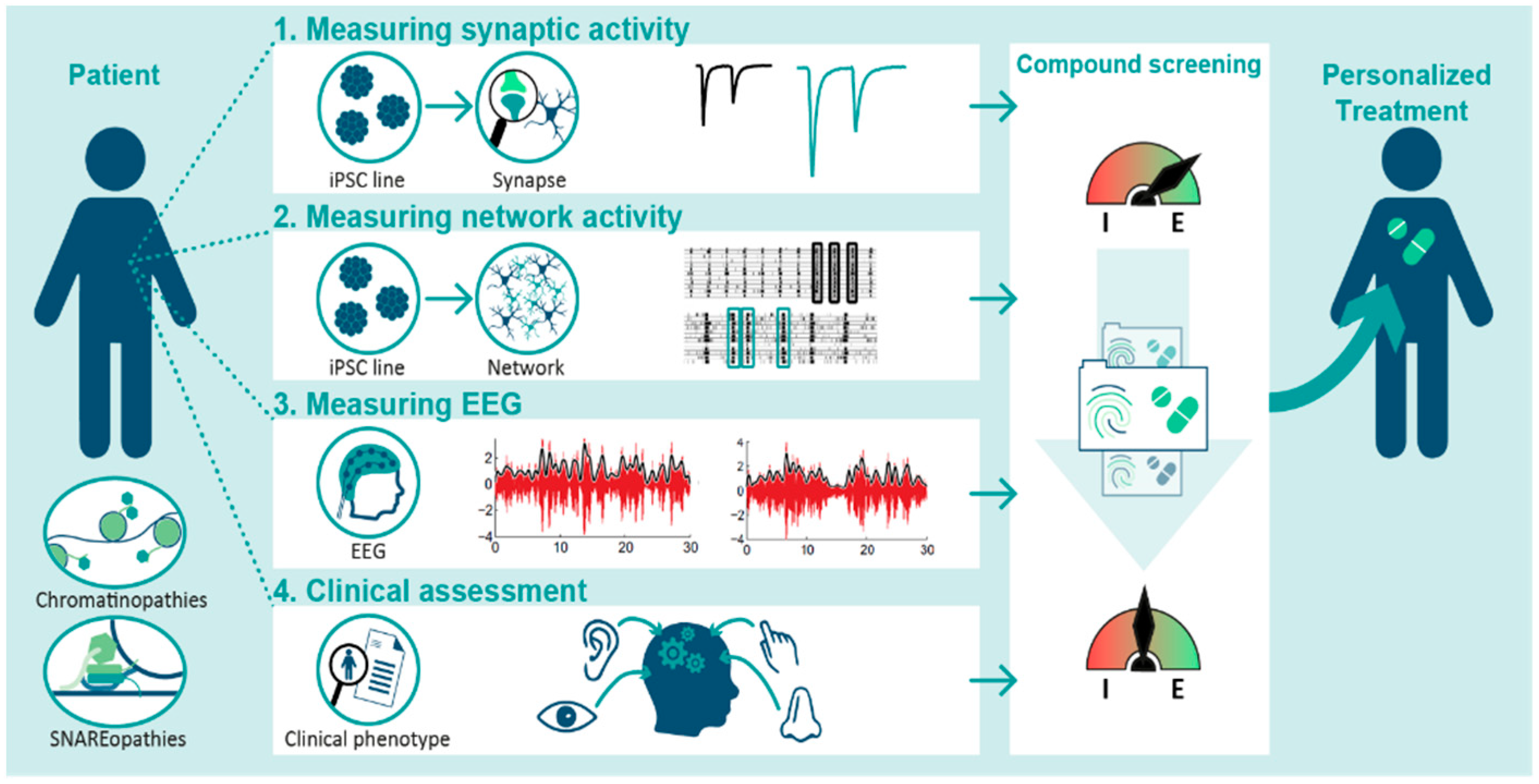
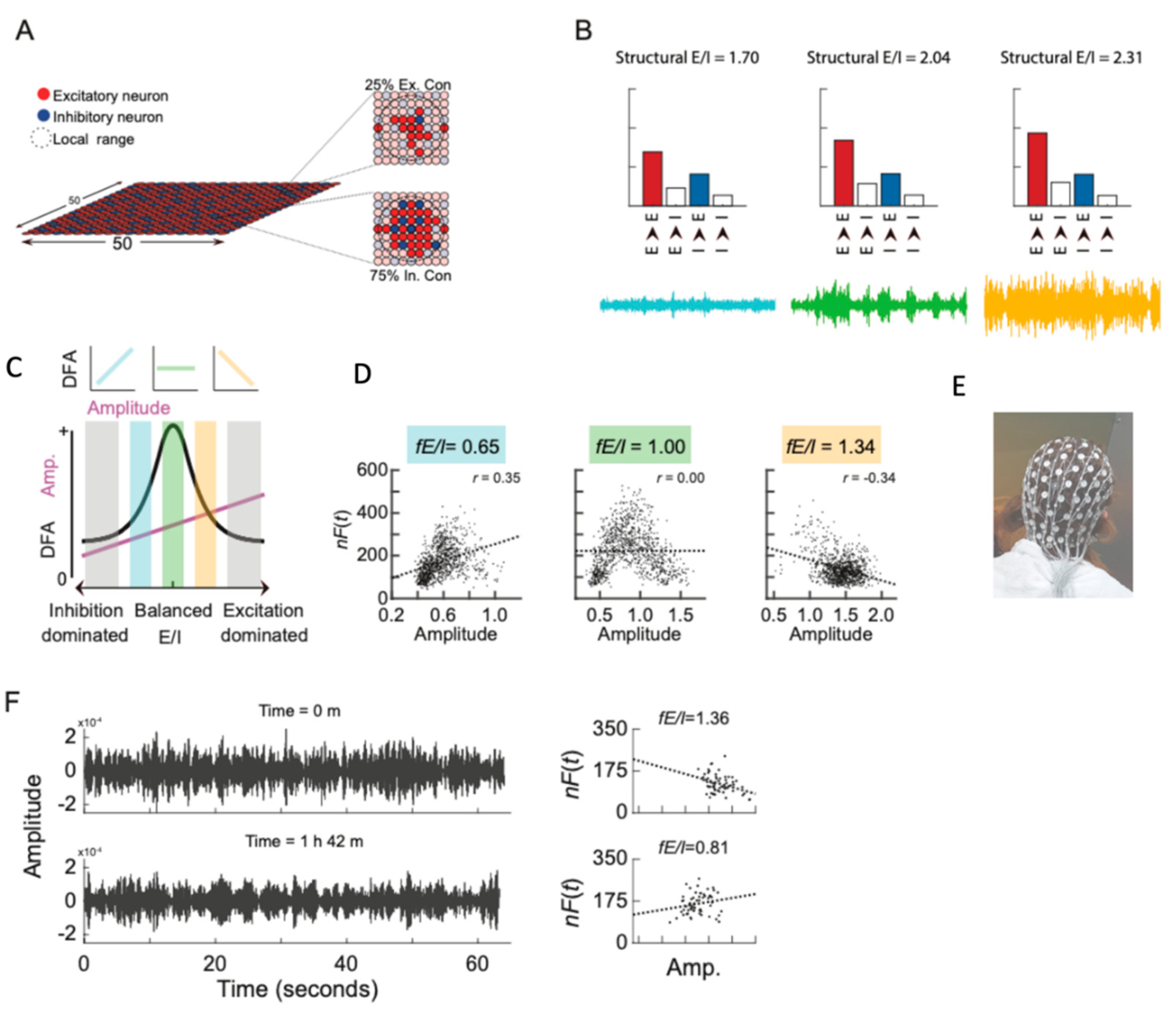
3. The Potential and Pitfalls of E/I Ratio Measurements in iPSC Models
4. Focus on Chromatinopathies and SNAREopathies
5. Connecting the Dots
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, Y.; Provenzano, G.; Casarosa, S. Neurobiological bases of autism-epilepsy comorbidity: A focus on excitation/inhibition imbalance. Eur. J. Neurosci. 2018, 47, 534–548. [Google Scholar] [CrossRef] [Green Version]
- Foss-Feig, J.H.; Adkinson, B.D.; Ji, J.L.; Yang, G.; Srihari, V.H.; McPartland, J.C.; Krystal, J.H.; Murray, J.D.; Anticevic, A. Searching for Cross-Diagnostic Convergence: Neural Mechanisms Governing Excitation and Inhibition Balance in Schizophrenia and Autism Spectrum Disorders. Biol. Psychiatry 2017, 81, 848–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeste, S.S.; Frohlich, J.; Loo, S.K. Electrophysiological biomarkers of diagnosis and outcome in neurodevelopmental disorders. Curr. Opin. Neurol. 2015, 28, 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naaijen, J.; Bralten, J.; Poelmans, G.; Glennon, J.C.; Franke, B.; Buitelaar, J.K. Glutamatergic and GABAergic gene sets in attention-deficit/hyperactivity disorder: Association to overlapping traits in ADHD and autism. Transl. Psychiatry 2017, 7, e999. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.B.; Valakh, V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selten, M.; van Bokhoven, H.; Kasri, N.N. Inhibitory control of the excitatory/inhibitory balance in psychiatric disorders. F1000Research 2018, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Rubenstein, J.L.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef]
- Isaacson, J.S.; Scanziani, M. How inhibition shapes cortical activity. Neuron 2011, 72, 231–243. [Google Scholar] [CrossRef] [Green Version]
- Antoine, M.W.; Langberg, T.; Schnepel, P.; Feldman, D.E. Increased Excitation-Inhibition Ratio Stabilizes Synapse and Circuit Excitability in Four Autism Mouse Models. Neuron 2019, 101, 648–661.e4. [Google Scholar] [CrossRef] [Green Version]
- Boutros, N. Epileptiform discharges in psychiatric patients: A controversy in need of resurrection. Clin. EEG Neurosci. 2009, 40, 239–244. [Google Scholar] [CrossRef]
- Bruining, H.; Hardstone, R.; Juarez-Martinez, E.L.; Sprengers, J.; Avramiea, A.E.; Simpraga, S.; Houtman, S.J.; Poil, S.S.; Dallares, E.; Palva, S.; et al. Measurement of excitation-inhibition ratio in autism spectrum disorder using critical brain dynamics. Sci. Rep. 2020, 10, 9195. [Google Scholar] [CrossRef]
- Spence, S.J.; Schneider, M.T. The role of epilepsy and epileptiform EEGs in autism spectrum disorders. Pediatr. Res. 2009, 65, 599–606. [Google Scholar] [CrossRef]
- Bruining, H.; Passtoors, L.; Goriounova, N.; Jansen, F.; Hakvoort, B.; De Jonge, M.; Poil, S.S. Paradoxical Benzodiazepine Response: A Rationale for Bumetanide in Neurodevelopmental Disorders? Pediatrics 2015, 136, e539–e543. [Google Scholar] [CrossRef] [Green Version]
- Sprengers, J.J.; Van Andel, D.M.; Zuithoff, N.P.; Keijzer-Veen, M.G.; Schulp, A.J.; Scheepers, F.E.; Lilien, M.R.; Oranje, B.; Bruining, H. Bumetanide for Core Symptoms of Autism Spectrum Disorder (BAMBI): A Single Center, Double-Blinded, Participant-Randomized, Placebo-Controlled, Phase Two, Superiority Trial. J. Am. Acad. Child Adolesc. Psychiatry 2020, 60, 865–876. [Google Scholar] [CrossRef]
- Van Andel, D.M.; Sprengers, J.J.; Oranje, B.; Scheepers, F.E.; Jansen, F.E.; Bruining, H. Effects of bumetanide on neurodevelopmental impairments in patients with tuberous sclerosis complex: An open-label pilot study. Mol. Autism 2020, 11, 30. [Google Scholar] [CrossRef]
- Charman, T.; Loth, E.; Tillmann, J.; Crawley, D.; Wooldridge, C.; Goyard, D.; Ahmad, J.; Auyeung, B.; Ambrosino, S.; Banaschewski, T.; et al. The EU-AIMS Longitudinal European Autism Project (LEAP): Clinical characterisation. Mol. Autism 2017, 8, 27. [Google Scholar] [CrossRef]
- Trobiani, L.; Meringolo, M.; Diamanti, T.; Bourne, Y.; Marchot, P.; Martella, G.; Dini, L.; Pisani, A.; De Jaco, A.; Bonsi, P. The neuroligins and the synaptic pathway in Autism Spectrum Disorder. Neurosci. Biobehav. Rev. 2020, 119, 37–51. [Google Scholar] [CrossRef]
- Marchetto, M.C.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Shcheglovitov, A.; Shcheglovitova, O.; Yazawa, M.; Portmann, T.; Shu, R.; Sebastiano, V.; Krawisz, A.; Froehlich, W.; Bernstein, J.A.; Hallmayer, J.F.; et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013, 503, 267–271. [Google Scholar] [CrossRef] [Green Version]
- Llamosas, N.; Arora, V.; Vij, R.; Kilinc, M.; Bijoch, L.; Rojas, C.; Reich, A.; Sridharan, B.; Willems, E.; Piper, D.R.; et al. SYNGAP1 Controls the Maturation of Dendrites, Synaptic Function, and Network Activity in Developing Human Neurons. J. Neurosci. 2020, 40, 7980–7994. [Google Scholar] [CrossRef]
- Paşca, S.P.; Portmann, T.; Voineagu, I.; Yazawa, M.; Shcheglovitov, A.; Paşca, A.M.; Cord, B.; Palmer, T.D.; Chikahisa, S.; Nishino, S.; et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 2011, 17, 1657–1662. [Google Scholar] [CrossRef]
- Sun, A.X.; Yuan, Q.; Fukuda, M.; Yu, W.; Yan, H.; Lim, G.G.Y.; Nai, M.H.; D’agostino, G.A.; Tran, H.D.; Itahana, Y.; et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science 2019, 366, 1486–1492. [Google Scholar] [CrossRef]
- Susco, S.G.; Arias-García, M.A.; López-Huerta, V.G.; Beccard, A.; Bara, A.M.; Moffitt, J.; Korn, J.; Fu, Z.; Barrett, L.E. FMR1 loss in a human stem cell model reveals early changes to intrinsic membrane excitability. Dev. Biol. 2020, 468, 93–100. [Google Scholar] [CrossRef]
- Becker, M.; Mastropasqua, F.; Reising, J.P.; Maier, S.; Ho, M.L.; Rabkina, I.; Li, D.; Neufeld, J.; Ballenberger, L.; Myers, L.; et al. Presynaptic dysfunction in CASK-related neurodevelopmental disorders. Transl. Psychiatry 2020, 10, 312. [Google Scholar] [CrossRef]
- Yi, F.; Danko, T.; Botelho, S.C.; Patzke, C.; Pak, C.; Wernig, M.; Südhof, T.C. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 2016, 352, aaf2669. [Google Scholar] [CrossRef] [Green Version]
- Dolmetsch, R.; Geschwind, D.H. The human brain in a dish: The promise of iPSC-derived neurons. Cell 2011, 145, 831–834. [Google Scholar] [CrossRef] [Green Version]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Chanda, S.; Marro, S.; Ng, Y.H.; Janas, J.A.; Haag, D.; Ang, C.E.; Tang, Y.; Flores, Q.; Mall, M.; et al. Generation of pure GABAergic neurons by transcription factor programming. Nat. Methods 2017, 14, 621–628. [Google Scholar] [CrossRef]
- Mossink, B.; Van Rhijn, J.R.; Wang, S.; Linda, K.; Vitale, M.R.; Zöller, J.E.; van Hugte, E.J.; Bak, J.; Verboven, A.H.; Selten, M.; et al. Cadherin-13 is a critical regulator of GABAergic modulation in human stem-cell-derived neuronal networks. Mol. Psychiatry 2021, 1–18. [Google Scholar] [CrossRef]
- Busskamp, V.; Lewis, N.E.; Guye, P.; Ng, A.H.; Shipman, S.L.; Byrne, S.M.; Sanjana, N.E.; Murn, J.; Li, Y.; Li, S.; et al. Rapid neurogenesis through transcriptional activation in human stem cells. Mol. Syst. Biol. 2014, 10, 760. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ward, M.E.; Chen, R.; Liu, K.; Tracy, T.E.; Chen, X.; Xie, M.; Sohn, P.D.; Ludwig, C.; Meyer-Franke, A.; et al. Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Rep. 2017, 9, 1221–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deneault, E.; Faheem, M.; White, S.H.; Rodrigues, D.C.; Sun, S.; Wei, W.; Piekna, A.; Thompson, T.; Howe, J.L.; Chalil, L.; et al. CNTN5-/+or EHMT2-/+human iPSC-derived neurons from individuals with autism develop hyperactive neuronal networks. eLife 2019, 8, e40092. [Google Scholar] [CrossRef]
- Meijer, M.; Rehbach, K.; Brunner, J.W.; Classen, J.A.; Lammertse, H.C.; van Linge, L.A.; Schut, D.; Krutenko, T.; Hebisch, M.; Cornelisse, L.N.; et al. A Single-Cell Model for Synaptic Transmission and Plasticity in Human iPSC-Derived Neurons. Cell Rep. 2019, 27, 2199–2211.e6. [Google Scholar] [CrossRef] [Green Version]
- Rhee, H.J.; Shaib, A.H.; Rehbach, K.; Lee, C.; Seif, P.; Thomas, C.; Gideons, E.; Guenther, A.; Krutenko, T.; Hebisch, M.; et al. An Autaptic Culture System for Standardized Analyses of iPSC-Derived Human Neurons. Cell Rep. 2019, 27, 2212–2228.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossink, B.; Verboven, A.H.; van Hugte, E.J.; Gunnewiek, T.M.K.; Parodi, G.; Linda, K.; Schoenmaker, C.; Kleefstra, T.; Kozicz, T.; van Bokhoven, H.; et al. Human neuronal networks on micro-electrode arrays are a highly robust tool to study disease-specific genotype-phenotype correlations in vitro. Stem Cell Rep. 2021, 16, 2182–2196. [Google Scholar] [CrossRef]
- Lin, H.C.; He, Z.; Ebert, S.; Schörnig, M.; Santel, M.; Nikolova, M.T.; Weigert, A.; Hevers, W.; Kasri, N.N.; Taverna, E. NGN2 induces diverse neuron types from human pluripotency. Stem Cell Rep. 2021, 16, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, R.; Koukouli, F.; Blanchard, S.; Catteau, J.; Raïs, C.; Lemonnier, T.; Féraud, O.; Bennaceur-Griscelli, A.; Groszer, M.; Maskos, U. Long-term development of human iPSC-derived pyramidal neurons quantified after transplantation into the neonatal mouse cortex. Dev. Biol. 2020, 461, 86–95. [Google Scholar] [CrossRef]
- Vitrac, A.; Pons, S.; Balkota, M.; Lemière, N.; Raïs, C.; Bourgeois, J.P.; Maskos, U.; Bourgeron, T.; Cloëz-Tayarani, I. A chimeric mouse model to study human iPSC-derived neurons: The case of a truncating SHANK3 mutation. Sci. Rep. 2020, 10, 13315. [Google Scholar] [CrossRef]
- Lavazza, A. Human cerebral organoids and consciousness: A double-edged sword. Monash Bioeth. Rev. 2020, 38, 105–128. [Google Scholar] [CrossRef] [PubMed]
- Lavazza, A. Potential ethical problems with human cerebral organoids: Consciousness and moral status of future brains in a dish. Brain Res. 2021, 1750, 147146. [Google Scholar] [CrossRef] [PubMed]
- Lammertse, H.C.A.; van Berkel, A.A.; Iacomino, M.; Toonen, R.F.; Striano, P.; Gambardella, A.; Verhage, M.; Zara, F. Homozygous STXBP1 variant causes encephalopathy and gain-of-function in synaptic transmission. Brain 2019, 143, 441–451. [Google Scholar] [CrossRef]
- Frega, M.; Linda, K.; Keller, J.M.; Gümüş-Akay, G.; Mossink, B.; van Rhijn, J.-R.; Negwer, M.; Gunnewiek, T.K.; Foreman, K.; Kompier, N.; et al. Neuronal network dysfunction in a model for Kleefstra syndrome mediated by enhanced NMDAR signaling. Nat. Commun. 2019, 10, 4928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef]
- Yahata, N.; Asai, M.; Kitaoka, S.; Takahashi, K.; Asaka, I.; Hioki, H.; Kaneko, T.; Maruyama, K.; Saido, T.C.; Nakahata, T.; et al. Anti-Aβ drug screening platform using human iPS cell-derived neurons for the treatment of Alzheimer’s disease. PLoS ONE 2011, 6, e25788. [Google Scholar] [CrossRef] [PubMed]
- Ciptasari, U.; van Bokhoven, H. The phenomenal epigenome in neurodevelopmental disorders. Hum. Mol. Genet. 2020, 29, R42–R50. [Google Scholar] [CrossRef] [PubMed]
- Kleefstra, T.; de Leeuw, N. Kleefstra Syndrome. In GeneReviews(®); Adam, M.P., Ed.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Gallagher, D.; Voronova, A.; Zander, M.A.; Cancino, G.I.; Bramall, A.; Krause, M.P.; Abad, C.; Tekin, M.; Neilsen, P.; Callen, D.; et al. Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Dev. Cell 2015, 32, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Kummeling, J.; Stremmelaar, D.E.; Raun, N.; Reijnders, M.R.F.; Willemsen, M.H.; Ruiterkamp-Versteeg, M.; Schepens, M.; Man, C.C.O.; Gilissen, C.; Cho, M.T.; et al. Characterization of SETD1A haploinsufficiency in humans and Drosophila defines a novel neurodevelopmental syndrome. Mol. Psychiatry 2021, 26, 2013–2024. [Google Scholar] [CrossRef]
- Adam, M.P.; Hudgins, L.; Hannibal, M. Kabuki syndrome. In GeneReviews(®); Adam, M.P., Ed.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Martens, M.B.; Frega, M.; Classen, J.; Epping, L.; Bijvank, E.; Benevento, M.; van Bokhoven, J.; Tiesinga, P.; Schubert, D.; Kasri, N.N. Euchromatin histone methyltransferase 1 regulates cortical neuronal network development. Sci. Rep. 2016, 6, 35756. [Google Scholar] [CrossRef] [Green Version]
- Negwer, M.; Piera, K.; Hesen, R.; Lütje, L.; Aarts, L.; Schubert, D.; Kasri, N.N. EHMT1 regulates Parvalbumin-positive interneuron development and GABAergic input in sensory cortical areas. Brain Struct. Funct. 2020, 225, 2701–2716. [Google Scholar] [CrossRef]
- Frega, M.; Selten, M.; Mossink, B.; Keller, J.M.; Linda, K.; Moerschen, R.; Qu, J.; Koerner, P.; Jansen, S.; Oudakker, A.; et al. Distinct Pathogenic Genes Causing Intellectual Disability and Autism Exhibit a Common Neuronal Network Hyperactivity Phenotype. Cell Rep. 2020, 30, 173–186.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhage, M.; Sorensen, J.B. SNAREopathies: Diversity in Mechanisms and Symptoms. Neuron 2020, 107, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Pintacuda, G.; Martín, J.M.; Eggan, K.C. Mind the translational gap: Using iPS cell models to bridge from genetic discoveries to perturbed pathways and therapeutic targets. Mol. Autism 2021, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Hardstone, R.; Poil, S.-S.; Schiavone, G.; Jansen, R.; Nikulin, V.V.; Mansvelder, H.D.; Linkenkaer-Hansen, K. Detrended fluctuation analysis: A scale-free view on neuronal oscillations. Front. Physiol. 2012, 3, 450. [Google Scholar] [CrossRef] [Green Version]
- Linkenkaer-Hansen, K.; Nikouline, V.V.; Palva, J.M.; Ilmoniemi, R. Long-range temporal correlations and scaling behavior in human brain oscillations. J. Neurosci. 2001, 21, 1370–1377. [Google Scholar] [CrossRef] [Green Version]
- Poil, S.-S.; de Haan, W.; van der Flier, W.M.; Mansvelder, H.D.; Scheltens, P.; Linkenkaer-Hansen, K. Integrative EEG biomarkers predict progression to Alzheimer’s disease at the MCI stage. Front. Aging Neurosci. 2013, 5, 58. [Google Scholar] [CrossRef] [Green Version]
- Poil, S.-S.; Hardstone, R.; Mansvelder, H.; Linkenkaer-Hansen, K. Critical-state dynamics of avalanches and oscillations jointly emerge from balanced excitation/inhibition in neuronal networks. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 9817–9823. [Google Scholar] [CrossRef]
- Beggs, J.M.; Plenz, D. Neuronal avalanches in neocortical circuits. J. Neurosci. 2003, 23, 11167–11177. [Google Scholar] [CrossRef] [Green Version]
- Poil, S.S.; van Ooyen, A.; Linkenkaer-Hansen, K. Avalanche dynamics of human brain oscillations: Relation to critical branching processes and temporal correlations. Hum. Brain Mapp. 2008, 29, 770–777. [Google Scholar] [CrossRef]
- van Andel, D.M.; van Stel, H.F.; Scheepers, F.E.; Oostrom, K.J.; Haverman, L.; Bruining, H. The sensory-reactivity PROM set: Identification of a parent reported outcome measure set for autism spectrum disorder. J. Patient Rep. Outcomes 2021, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Schauder, K.B.; Bennetto, L. Toward an Interdisciplinary Understanding of Sensory Dysfunction in Autism Spectrum Disorder: An Integration of the Neural and Symptom Literatures. Front. Neurosci. 2016, 10, 268. [Google Scholar] [CrossRef] [PubMed]
- Wigham, S.; Rodgers, J.; South, M.; McConachie, H.; Freeston, M. The interplay between sensory processing abnormalities, intolerance of uncertainty, anxiety and restricted and repetitive behaviours in autism spectrum disorder. J. Autism Dev. Disord. 2015, 45, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Guidance for Industry: Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labelling Claims. Available online: www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM193282.pdf (accessed on 5 December 2020).
- Streiner, D.L.; Norman, G.R. Health Measure Scales: A Practical Guide to Their Development and Use, 4th ed.; Oxford University Press: New York, NY, USA, 2008. [Google Scholar]
- Cella, D.; Yount, S.; Rothrock, N.; Gershon, R.; Cook, K.; Reeve, B.; Ader, D.; Fries, J.F.; Bruce, B.; Rose, M. The Patient-Reported Outcomes Measurement Information System (PROMIS): Progress of an NIH Roadmap cooperative group during its first two years. Med. Care 2007, 45, S3–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- HealthMeasures. Computer Adaptive Tests (CATs). Available online: https://www.healthmeasures.net/resource-center/measurement-science/computer-adaptive-tests-cats (accessed on 26 February 2021).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geertjens, L.; van Voorst, T.W.; Bouman, A.; van Boven, M.A.; Kleefstra, T.; Verhage, M.; Linkenkaer-Hansen, K.; Nadif Kasri, N.; Cornelisse, L.N.; Bruining, H. Following Excitation/Inhibition Ratio Homeostasis from Synapse to EEG in Monogenetic Neurodevelopmental Disorders. Genes 2022, 13, 390. https://doi.org/10.3390/genes13020390
Geertjens L, van Voorst TW, Bouman A, van Boven MA, Kleefstra T, Verhage M, Linkenkaer-Hansen K, Nadif Kasri N, Cornelisse LN, Bruining H. Following Excitation/Inhibition Ratio Homeostasis from Synapse to EEG in Monogenetic Neurodevelopmental Disorders. Genes. 2022; 13(2):390. https://doi.org/10.3390/genes13020390
Chicago/Turabian StyleGeertjens, Lisa, Torben W. van Voorst, Arianne Bouman, Maaike A. van Boven, Tjitske Kleefstra, Matthijs Verhage, Klaus Linkenkaer-Hansen, Nael Nadif Kasri, L. Niels Cornelisse, and Hilgo Bruining. 2022. "Following Excitation/Inhibition Ratio Homeostasis from Synapse to EEG in Monogenetic Neurodevelopmental Disorders" Genes 13, no. 2: 390. https://doi.org/10.3390/genes13020390
APA StyleGeertjens, L., van Voorst, T. W., Bouman, A., van Boven, M. A., Kleefstra, T., Verhage, M., Linkenkaer-Hansen, K., Nadif Kasri, N., Cornelisse, L. N., & Bruining, H. (2022). Following Excitation/Inhibition Ratio Homeostasis from Synapse to EEG in Monogenetic Neurodevelopmental Disorders. Genes, 13(2), 390. https://doi.org/10.3390/genes13020390